
Stochasticity Highlights the Development of Both the Gastrointestinal and Upper-Respiratory-Tract Microbiomes of Neonatal Dairy Calves in Early Life

 , , and
, , and
Simple Summary
Abstract
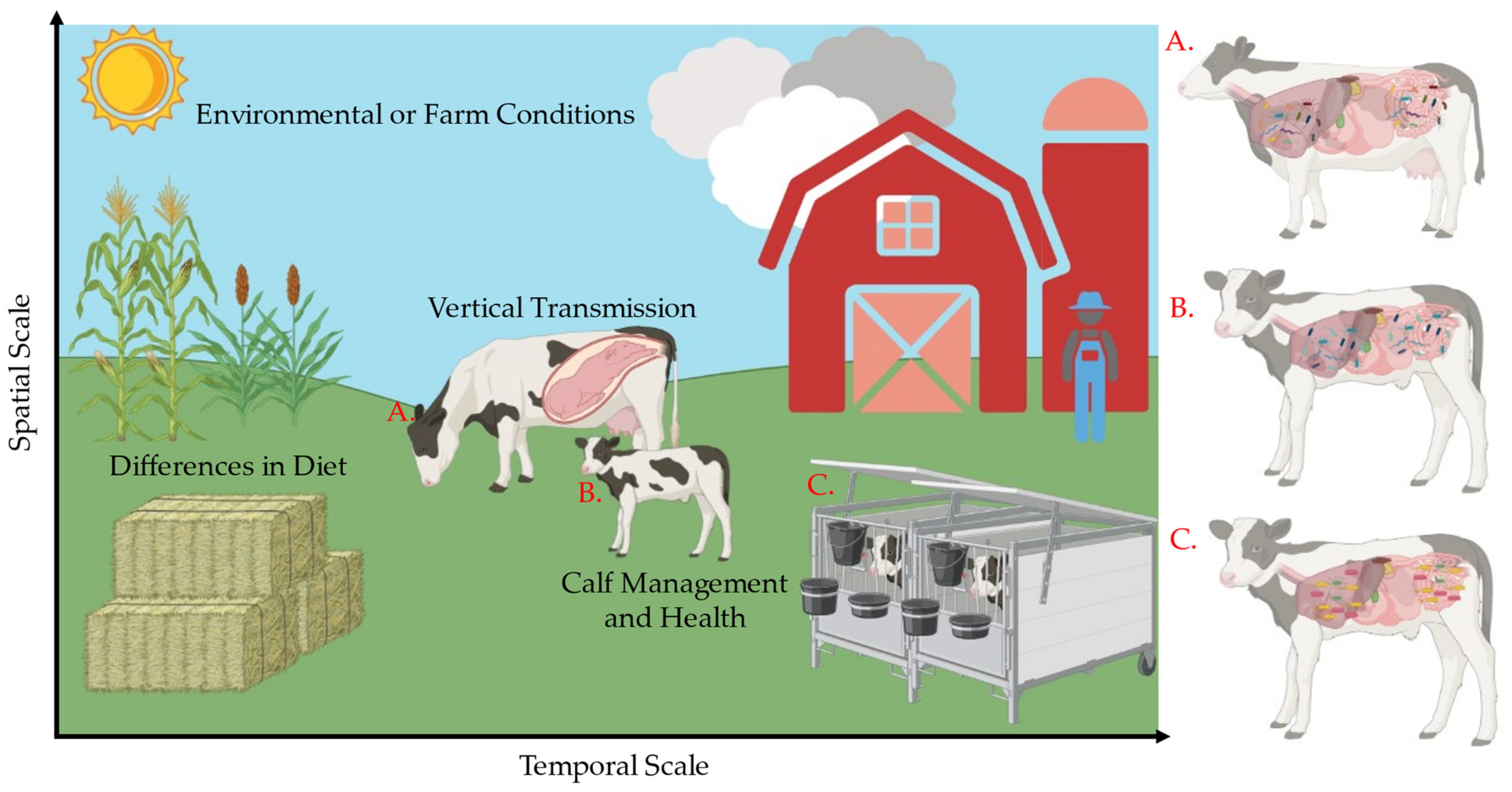
1. Introduction
2. Materials and Methods
2.1. Animals, Facilities, and Experimental Design
2.2. Microbial Sampling and Clinical Health Observations
2.3. DNA Extraction and Sequencing
2.4. Analysis of Fecal and Nasal Microbiomes
3. Results
3.1. Calf Characteristics and Sequencing Results
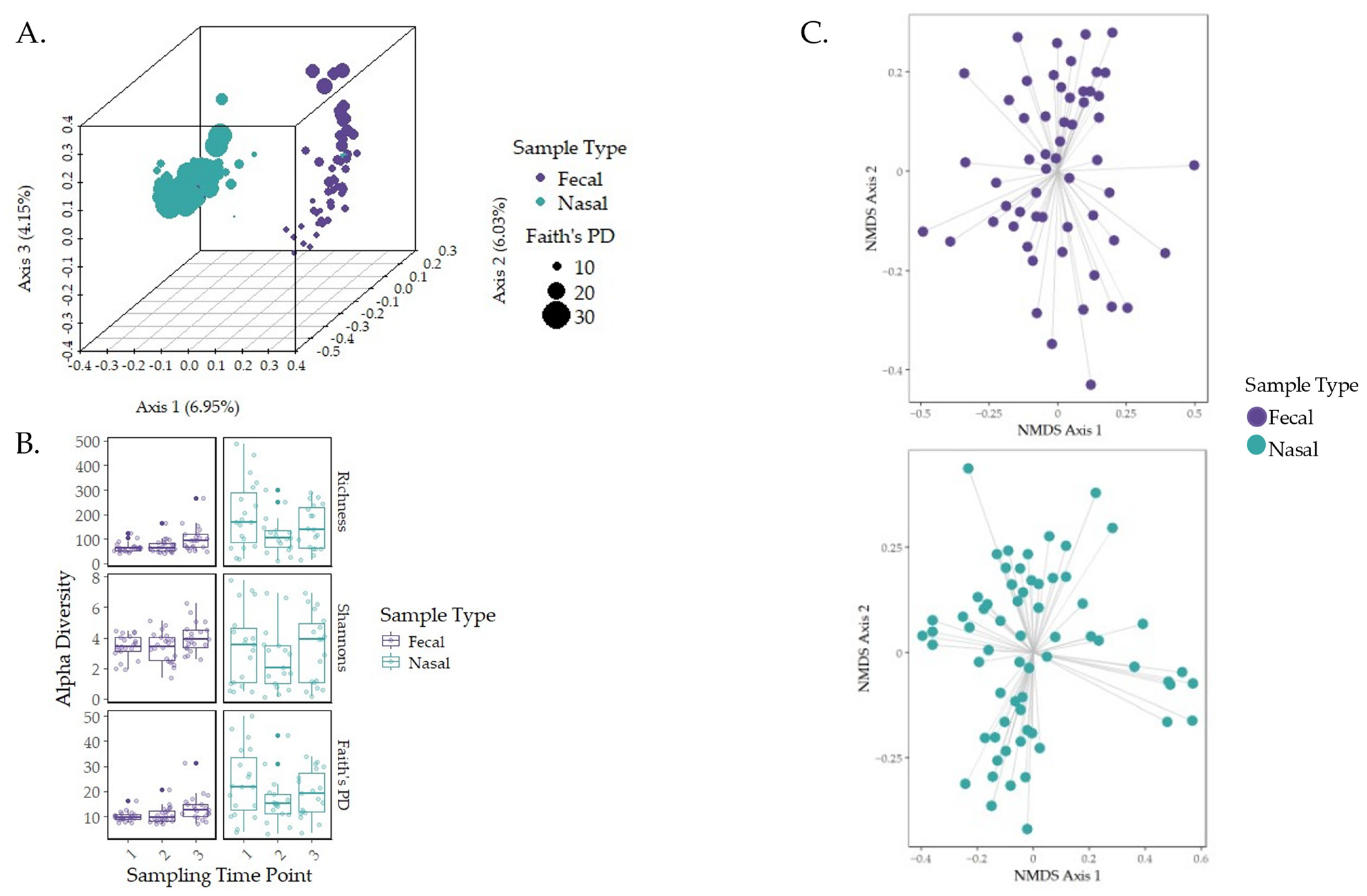
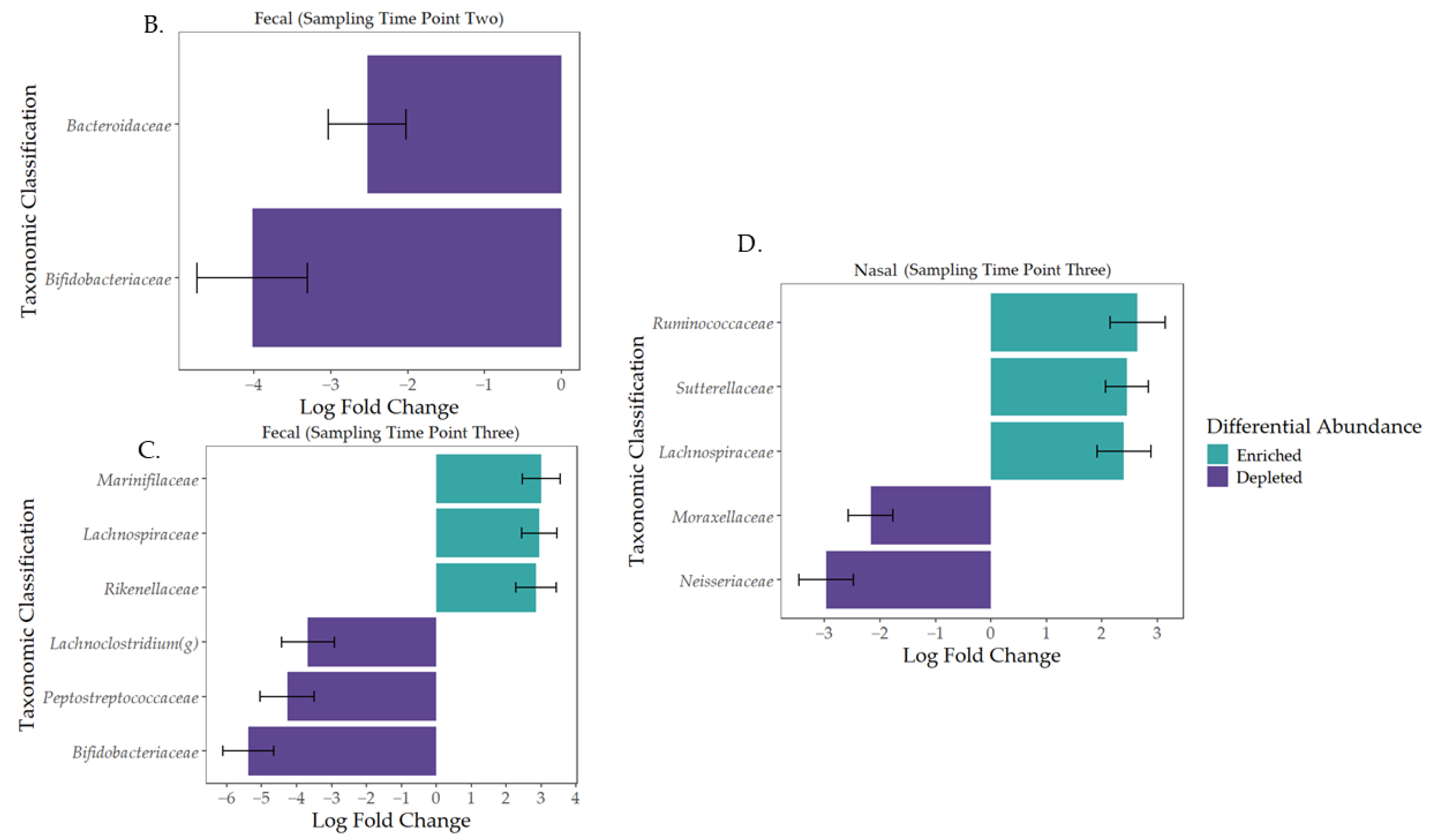
3.2. Characterization of the Early-Life Microbiome
3.3. Governing Forces of Microbial Community Composition
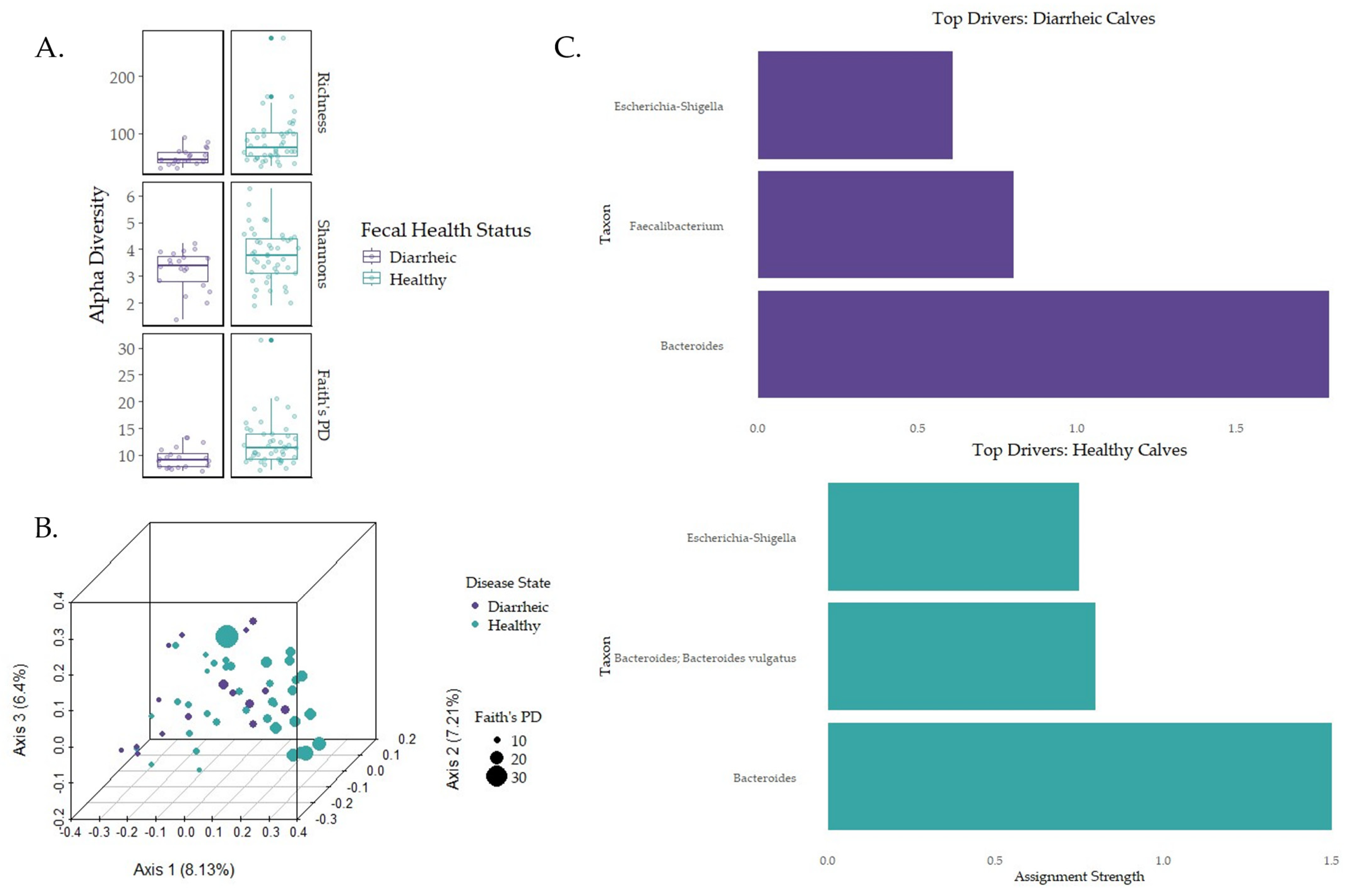
3.4. Impacts of Disease on Early-Life Microbial Establishment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKP | Anna Karenina Principles |
| ANCOM-BC | Analysis of composition of microbiomes with bias corrections |
| BRD | Bovine respiratory disease |
| DEMENT | Decomposition Model of Enzymatic Traits |
| DMM | Dirichlet multinomial model |
| FDR | False discovery rate |
| GI | Gastrointestinal |
| NMDS | Nonmetric multidimensional |
| NST | Normalized stochasticity testing |
| NSTi | Normalized stochasticity index |
| MST | Modified stochasticity ratio |
| PERMANOVA | Permutational multivariant analysis of variance |
| βRC | Raup–Crick distance |
| SES | Standardized effect size index |
| URT | Upper respiratory tract |
References
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Loso, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, S.; Shen, N.; Wu, H.C.; Clemente, J.C. The microbiome in early-life: Implications for health outcomes. Nat. Med. 2016, 22, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Mohammadkah, A.I.; Simpson, E.B.; Patterson, S.G.; Ferguson, J.F. Development of the gut microbiome in children, and lifetime implications for obesity and cardiometabolic disease. Children 2018, 5, 160. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Chen, H.; Zhang, S.; Zhuang, J.; Li, Q.; Feng, Z. Intestinal microbiota in early-life and its implications on childhood health. Gen. Prot. Bioinform. 2019, 17, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Anny. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewaski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The impact of gut microbiota on brain and behaviour: Implications for psychiatry. Cur. Opi. Clin. Nutr. Metabl. Care 2015, 18, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.V.-A.; Foster, K.R. Why does the microbiome affect behaviour? Nat. Rev. Microbiol. 2018, 16, 647–655. [Google Scholar] [CrossRef] [PubMed]
- de Menezes, A.B.; Lewis, E.; O’Donovan, M.; O’Neail, B.F.; Clipson, N.; Doyle, E.M. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol. Ecol. 2011, 78, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.-D.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 2018, 14, e10007580. [Google Scholar] [CrossRef]
- Chen, S.; Wang, J.; Peng, D.; Li, G.; Chen, J.; Gu, X. Exposure to heat-stress environment affects the physiology, circulation levels of cytokines, and the microbiome in dairy cows. Sci. Rep. 2018, 8, 14606. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, E.; Kelly, A.; McCabe, M.S.; Kenny, D.A.; Guan, L.L.; Waters, S.M. Effect of butyrate-fortified milk replacer on gastrointestinal microbiota and products of fermentation in artificially reared dairy calves at weaning. Sci. Rep. 2018, 8, 14901. [Google Scholar] [CrossRef] [PubMed]
- Bronzo, V.; Lopreiato, V.; Riva, F.; Amadori, M.; Curone, G.; Addis, M.F.; Cremonesi, P.; Moroni, P.; Trevisi, E.; Castiglioni, B. The role of innate immune response and microbiome in resilience of dairy cattle to disease: The mastitis model. Animals 2020, 10, 1397. [Google Scholar] [CrossRef] [PubMed]
- Perez-Muñoz, M.E.; Arrieta, M.-C.; Ramer-Tait, A.E.; Walter, J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: Implications for research on the pioneer infant microbiome. Microbiome 2017, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.X.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1904. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2013, 166, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Takino, T.; Kato-Mori, Y.; Motooka, D.; Nakamura, S.; Iida, T.; Hagiwara, K. Postnatal changes in the relative abundance of intestinal Lactobacillus spp. in newborn calves. J. Vet. Med. Sci. 2017, 79, 452–455. [Google Scholar] [CrossRef]
- Haley, B.J.; Kim, S.-W.; Salaheen, S.; Hovingh, E.; Van Kessel, J.A.S. Differences in the microbial community and resistome structures of feces from preweaned calves and lactating cows in commercial dairy herds. Food. Path. Dis. 2020, 17, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.N.; Beck, M.R.; Waldrip, H.; Koziel, J.A. Connecting the ruminant microbiome to climate change: Insights from current ecological and evolutionary concepts. Front. Microbiol. 2024, 15, 1503315. [Google Scholar] [CrossRef] [PubMed]
- Vellend, M.; Srivastava, D.S.; Anderson, K.M.; Brown, C.D.; Jankowski, J.E.; Kleynhans, E.J.; Kraft, N.J.B.; Letaw, A.D.; MacDonald, A.A.M.; Myers-Smith, I.H.; et al. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 2014, 123, 1420–1430. [Google Scholar] [CrossRef]
- Evans, S.; Martiny, J.B.H.; Allison, S.D. Effects of dispersal and selection on stochastic assembly in microbial communities. ISME J. 2016, 11, 146–185. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Ma, T.; Steele, M.; Guan, L.L. Varied microbial community assembly and specialization patterns driven by early life microbiome perturbation and modulation in young ruminants. ISME J. 2024, 4, ycae044. [Google Scholar] [CrossRef]
- USDA. Part II: Changes in the U.S. Dairy Cattle industry, 1991–2007. In Dairy 2007; USDA-APHIS-VS, CEAH: Fort Collins, CO, USA, 2007; pp. 57–61. [Google Scholar]
- USDA. Part IV: Health and health management of U.S. feedlots with a capacity of 1000 or more head. In Feedlot 2011; APHIS, National Animal Health Monitoring System: Fort Collins, CO, USA, 2013. [Google Scholar]
- Dunn, T.R.; Ollivett, T.L.; Renaud, D.L.; Leslie, K.E.; LeBlanc, S.J.; Duffield, T.F.; Kelton, D.F. The effect of lung consolidation, as determined by ultrasonography, on first-lactation milk production in Holstein dairy calves. J. Dairy Sci. 2018, 101, 5404–5410. [Google Scholar] [CrossRef] [PubMed]
- Cramer, M.C.; Ollivett, T.L. Growth of preweaned, group-housed dairy calves diagnosed with respiratory disease using clinical respiratory scoring and thoracic ultrasound—A cohort study. J. Dairy Sci. 2019, 102, 4322–4331. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.E.; Arroyo, L.G.; Costa, M.C.; Viel, L.; Weese, J.S. Characterization of the fecal bacterial microbiota of healthy and diarrheic dairy calves. J. Vet. Intern. Med. 2017, 31, 928–939. [Google Scholar] [CrossRef]
- Kim, E.-T.; Lee, S.-J.; Kim, T.-Y.; Lee, H.-G.; Atikur, R.M.; Gu, B.-H.; Kim, D.-H.; Park, B.-Y.; Son, J.-K.; Kim, M.-H. Dynamic changes in fecal microbial communities of neonatal dairy calves by aging and diarrhea. Animals 2021, 11, 1113. [Google Scholar] [CrossRef]
- Lima, S.F.; Teixeira, A.G.V.; Higgins, C.H.; Lima, F.S.; Bicalho, R.C. The upper respiratory tract microbiome and its potential role in bovine respiratory disease and otitis media. Sci. Rep. 2016, 6, 29050. [Google Scholar] [CrossRef] [PubMed]
- Centeno-Martinez, R.E.; Klopp, R.N.; Koziol, J.; Boerman, J.P.; Johnson, T.A. Dynamics of the nasopharyngeal microbiome of apparently healthy calves and those with clinical symptoms of bovine respiratory disease from disease diagnosis to recovery. Front. Vet. Sci. 2023, 10, 1297158. [Google Scholar] [CrossRef]
- Frazier, A.N.; Belk, A.D.; Beck, M.R.; Koziel, J.A. Impact of methane mitigation strategies on the native ruminant microbiome: A protocol for a systematic review and meta-analysis. PLoS ONE 2024, 19, e0308914. [Google Scholar] [CrossRef] [PubMed]
- Ferree, L.A.; Edwards-Callaway, L.N.; Ramon-Muniz, I.N.; Coetzee, J.F.; Applegate, T.J.; Ollivett, T.L.; Cramer, M.C. Oral meloxicam given as an ancillary treatment at respiratory disease diagnosis was not associated with growth, clinical scores, or ultrasound scores in preweaned dairy calves. J. Am. Vet. Med. Assoc. 2022, 261, 1716–1723. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.M.; Peek, S.F. Timely diagnosis of dairy calf respiratory disease using a standardized scoring system. Anim. Health Res. Rev. 2014, 15, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Ollivett, T.L.; Buczinski, S. On-farm use of ultrasonography for bovine respiratory disease. Vet. Clin. N. Am. Food Anim. Pract. 2016, 32, 19–35. [Google Scholar] [CrossRef]
- Belk, A.D.; Frazier, A.N.; Fuerniss, L.K.; Delmore, R.; Belk, K.; Borlee, B.; Geornaras, I.; Martin, J.N.; Metcalf, J.L. A Pilot Study: The development of a facility-associated microbiome and its association with the presence of Listeria spp. in one small meat processing facility. Microbiol. Spectr. 2022, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. The Earth Microbiome Project Consortium. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Navas-Molina, J.A.; Kosciolek, T.; McDonald, D.; Vázquez-Baeza, Y.; Ackermann, G.; DeReus, J.; Janssen, S.; Swafford, A.D.; Orchanian, S.B.; et al. Qiita: Rapid, web-enabled microbiome meta-analysis. Nat. Methods 2018, 15, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Boylen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 87, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Bokulich, N.; Dillon, M.; Bolyen, E.; Kaehler, B.D.; Huttley, G.A.; Caporaso, J.G. Q2-sample-classifier: Machine-learning tools for microbiome classification and regression. J. Open Source Softw. 2018, 3, 934. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11, 538. [Google Scholar] [CrossRef] [PubMed]
- Janssen, S.; McDonald, D.; Gonzalez, A.; Navas-Molina, J.A.; Jiang, L.; Xu, Z.Z.; Winker, K.; Kado, D.M.; Orwoll, E.; Manary, M.; et al. Phylogenetic placement of exact amplicon sequences improves associations with clinical information. mSystems 2018, 3, 3. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Das Peddada, S. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.L.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1949; Volume vii, p. 117. [Google Scholar]
- McIntosh, R.P. An Index of Diversity and the Relation of Certain Concepts to Diversity. Ecology 1967, 48, 392–404. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 260, 583–621. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing: Vienna, Austria. Available online: https://www.R-project.org (accessed on 10 April 2021).
- Zhou, J.; Ning, D. Stochastic community assembly: Does it matter in microbial ecology? Microbiol. Mol. Biol. R. 2017, 81, e00002-17. [Google Scholar] [CrossRef] [PubMed]
- Gotelli, N.J.; McCabe, D.J. Species co-occurrence: A meta-analysis of J.M. Diamond’s assembly rules model. Ecology 2002, 83, 2091–2096. [Google Scholar] [CrossRef]
- Crump, B.C.; Peterson, B.J.; Raymond, P.A.; Holmes, R.M. Circumpolar synchrony in big river bacterioplankton. Proc. Natl. Acad. Sci. USA 2009, 106, 21208–21212. [Google Scholar] [CrossRef]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef]
- Stewart, C.J.; Ajami, N.J.; O’Brien, J.L.; Hutchinson, D.S.; Smith, D.P.; Wong, M.C.; Ross, M.C.; Lloyd, R.E.; Doddapaneni, H.; Metcalf, G.A.; et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 2019, 562, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. R Package, Version 2.6-2; Vegan: Community Ecology Package; R Core Team: Vienna, Austria, 2022. Available online: https://CRAN.R-project.org/package=vegan (accessed on 9 January 2025).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Frazier, A.N. Application of Biotechnology in Agriculture: A bioinformatic and Market Analysis of Novel Intervention Methods. Ph.D. Thesis, Colorado State University, Fort Collins, CO, USA, 2020. Available online: https://hdl.handle.net/10217/234261 (accessed on 19 January 2025).
- Ma, T.; Villot, C.; Renaud, D.; Skidmore, A.; Chevaux, E.; Steele, M.; Guan, L.L. Linking perturbations to temporal changes in diversity, stability, and compositions of neonatal calf gut microbiota: Prediction of diarrhea. ISME J. 2020, 14, 2223–2235. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, D.; Zhang, Y.; Li, K.; Wang, M.; Ma, J. Dynamic distribution of gut microbiota in cattle at different breeds and health states. Front. Microbiol. 2023, 14, 1113730. [Google Scholar] [CrossRef]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Guan, L.L. Understanding the gut microbiome of dairy calves: Opportunities to improve early-life gut health. J. Dairy Sci. 2017, 100, 5996–6005. [Google Scholar] [CrossRef]
- Kelly, W.J.; Cookson, A.L.; Altermann, E.; Lambie, S.C.; Perry, R.; The, K.H.; Otter, D.E.; Shapiro, N.; Woyke, T.; Leahy, S.C. Genomic analysis of three Bifidobacterium species isolated from the calf gastrointestinal tract. Sci. Rep. 2016, 6, 30768. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tube, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect enteropathogenic infection through the production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef] [PubMed]
- O’Connell Motherway, M.; Zomer, A.; Leahy, S.C.; Reunanen, J.; Bottacini, F.; Claesson, M.J.; O’Brien, F.; Flynn, K.; Casey, P.G.; Munoz, J.A.M.; et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proc. Natl. Acad. Sci. USA 2011, 108, 11217–11222. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.-I.; Kim, Y.H.; Takashima, K.; Kimura, A.; Nagai, K.; Ichijo, T.; Sato, S. Composition of the microbiota in forestomach fluids and feces of Japanese Black calves with white scours. J. Anim. Sci. 2017, 95, 3949–3960. [Google Scholar] [CrossRef] [PubMed]
- Zanevald, J.R.; McMinds, R.; Thurber, R.V. Stress and stability: Applying the Anna Karenia principle to animal microbiomes. Nat. Microbiol. 2017, 2, 17121. [Google Scholar] [CrossRef] [PubMed]
- Xun, W.; Li, W.; Xiong, W.; Ren, Y.; Liu, Y.; Miao, Y.; Xu, Z.; Zhang, N.; Shen, Q.; Zhang, R. Diversity-triggered deterministic bacterial assembly constraints community functions. Nat. Commun. 2019, 10, 3833. [Google Scholar] [CrossRef]
- Appert, O.; Garcia, A.R.; Frei, R.; Roduit, C.; Constancias, F.; Neuzil-Bunesova, V.; Ferstl, R.; Zhang, J.; Akdis, C.; Lauener, R.; et al. Initial butyrate producers during infant gut microbiota development are endospore formers. Environ. Microbiol. 2020, 22, 3909–3921. [Google Scholar] [CrossRef]
- Lima, S.F.; de Souza Bicalha, M.L.; Bicalho, R.C. The Bos taurus maternal microbiome: Role in determining the progeny early-life upper respiratory tract microbiome and health. PLoS ONE 2018, 14, e0208014. [Google Scholar] [CrossRef] [PubMed]
- Centeno-Martinez, R.E.; Glidden, N.; Mohan, S.; Davidson, J.L.; Fernández-Juricic, E.; Boerman, J.P.; Schoonmaker, J.; Pillai, D.; Koziol, J.; Ault, A.; et al. Identification of bovine respiratory disease through the nasal microbiome. Anim. Microbiome 2022, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhang, S.; Xiang, Z.; Shirani, I.; Chen, Y.; Guo, A. Effects of vaccination and interventions on nasal microbiome and BRD-associated pathogens in calves. Front. Microbiol. 2024, 15, 1467908. [Google Scholar] [CrossRef] [PubMed]
- Barden, M.; Richards-Rios, P.; Ganda, E.; Lenzi, L.; Eccles, R.; Neary, J.; Oultram, J.; Oikonomou, G. Maternal influences on oral and faecal microbiota maturation in neonatal calves in beef and dairy production systems. Anim. Microbiome 2020, 2, 31. [Google Scholar] [CrossRef]
- Raabis, S.M.; Quick, A.E.; Skarlupka, V.J.H.; Suen, G.; Ollivett, T.L. The nasopharyngeal microbiota of preweaned dairy calves with and without ultrasonographic lung lesions. J. Dairy Sci. 2021, 104, 3386–3402. [Google Scholar] [CrossRef] [PubMed]
- Amat, S.; Timsit, E.; Workentine, M.; Schwinghamer, T.; van der Meer, F.; Guo, Y.; Alexander, T.W. A single intranasal dose of bacterial therapeutics to calves confers longitudinal modulation of the nasopharyngeal micreobiota: A pilot study. Msystems 2023, 8, e01016-22. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.D. A trait-based approach for modelling microbial litter decomposition. Ecol. Lett. 2012, 15, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Pitta, D.W.; Indugu, N.; Kumar, S.; Vecchiarelli, B.; Sinha, R.; Baker, L.D.; Bhukya, B.; Ferguson, J.D. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 2016, 38, 50–60. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Health Category * | Time Point 1 | Time Point 2 | Time Point 3 |
|---|---|---|---|
| Healthy | 16 | 10 | 14 |
| Breed | |||
| Jersey | 7 | 3 | 7 |
| Holstein | 6 | 5 | 5 |
| Cross | 3 | 2 | 2 |
| Sex | |||
| Male | 9 | 4 | 8 |
| Female | 7 | 6 | 6 |
| Diarrhea | 3 | 8 | 4 |
| Breed | |||
| Jersey | 2 | 5 | 2 |
| Holstein | 1 | 2 | 2 |
| Cross | 0 | 1 | 0 |
| Sex | |||
| Male | 2 | 6 | 2 |
| Female | 1 | 2 | 2 |
| BRD ¥ | 0 | 0 | 0 |
| BRD/Diarrhea | 0 | 1 ¶ | 1 ! |
| Sample Type | NSTi | MST | SES | βRC | |
|---|---|---|---|---|---|
| Fecal | Sampling time point | 0.3382 * (0.5411) | 0.3202 (0.5411) | 0.0482 (0.2185) | 0.0546 (0.2185) |
| Disease state | 0.4595 (0.6127) | 0.1415 (0.3774) | 0.9219 (0.9361) | 0.9361 (0.9361) | |
| Nasal ¥ | Sampling time point | 0.8345 (0.8344) | 0.6316 (0.8344) | 0.7071 (0.8344) | 0.7671 (0.8344) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frazier, A.N.; Ferree, L.; Belk, A.D.; Al-Lakhen, K.; Cramer, M.C.; Metcalf, J.L. Stochasticity Highlights the Development of Both the Gastrointestinal and Upper-Respiratory-Tract Microbiomes of Neonatal Dairy Calves in Early Life. Animals 2025, 15, 361. https://doi.org/10.3390/ani15030361
Frazier AN, Ferree L, Belk AD, Al-Lakhen K, Cramer MC, Metcalf JL. Stochasticity Highlights the Development of Both the Gastrointestinal and Upper-Respiratory-Tract Microbiomes of Neonatal Dairy Calves in Early Life. Animals. 2025; 15(3):361. https://doi.org/10.3390/ani15030361
Chicago/Turabian StyleFrazier, A. Nathan, Logan Ferree, Aeriel D. Belk, Khalid Al-Lakhen, M. Caitlin Cramer, and Jessica L. Metcalf. 2025. "Stochasticity Highlights the Development of Both the Gastrointestinal and Upper-Respiratory-Tract Microbiomes of Neonatal Dairy Calves in Early Life" Animals 15, no. 3: 361. https://doi.org/10.3390/ani15030361
APA StyleFrazier, A. N., Ferree, L., Belk, A. D., Al-Lakhen, K., Cramer, M. C., & Metcalf, J. L. (2025). Stochasticity Highlights the Development of Both the Gastrointestinal and Upper-Respiratory-Tract Microbiomes of Neonatal Dairy Calves in Early Life. Animals, 15(3), 361. https://doi.org/10.3390/ani15030361