
Genome Wide Identification of Structure Variations in Five Italian Turkey Populations

 ,
,  , ,
, ,  ,
,  ,
,  , ,
, ,  and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
2.2. Sequencing and Mapping
2.3. Analysis of Population Structure
2.4. SVs Calling and Annotation
2.5. Post-Filtration and SV Processing
2.6. Functional Annotation
2.7. Gene Ontology (GO) Terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
3. Results
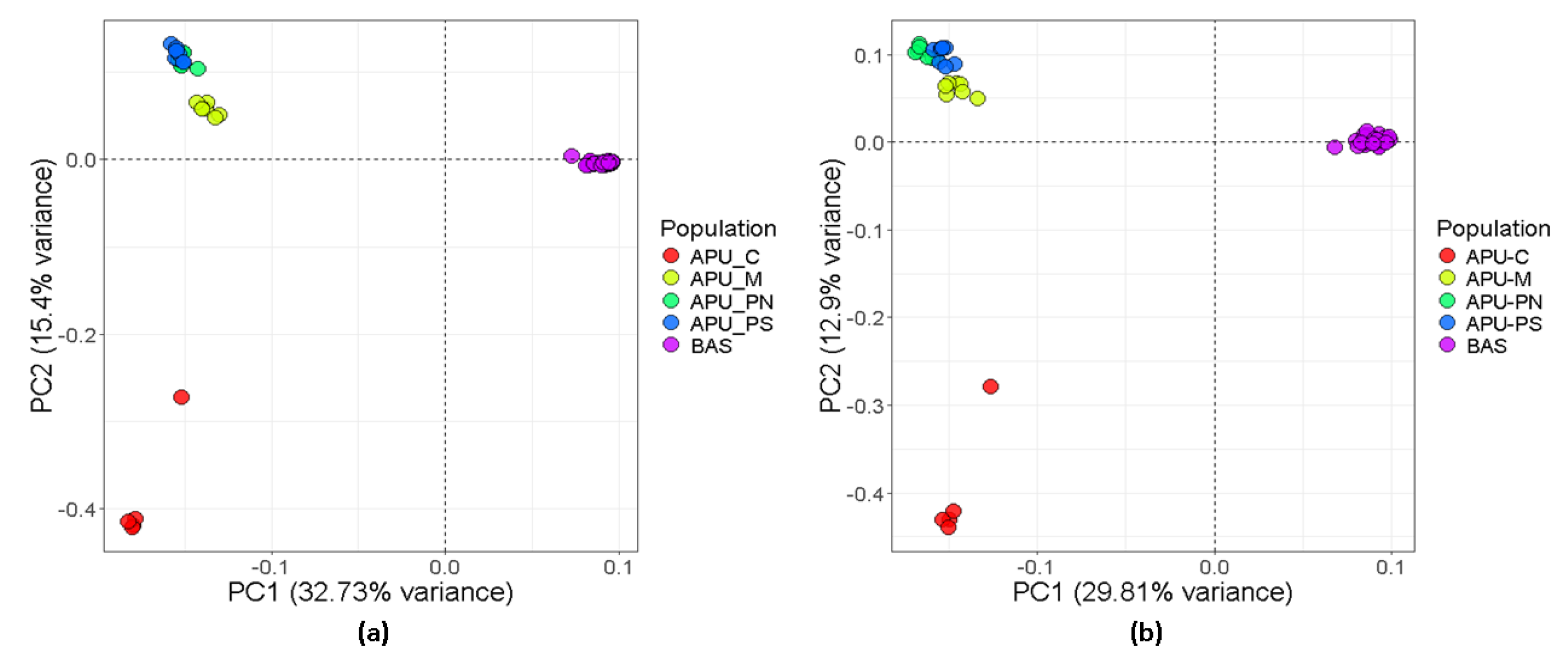
3.1. Population Genetic Structure
3.2. SV Discovery and Characterization
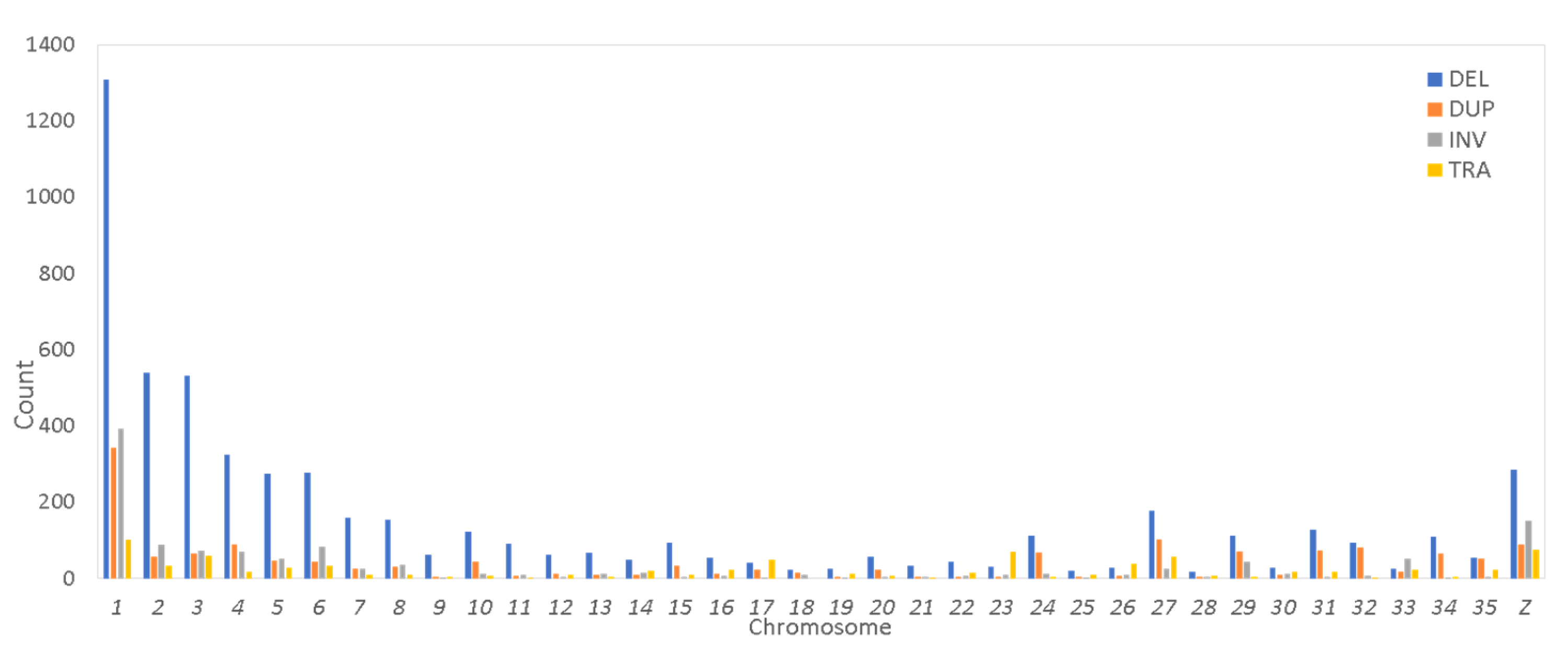
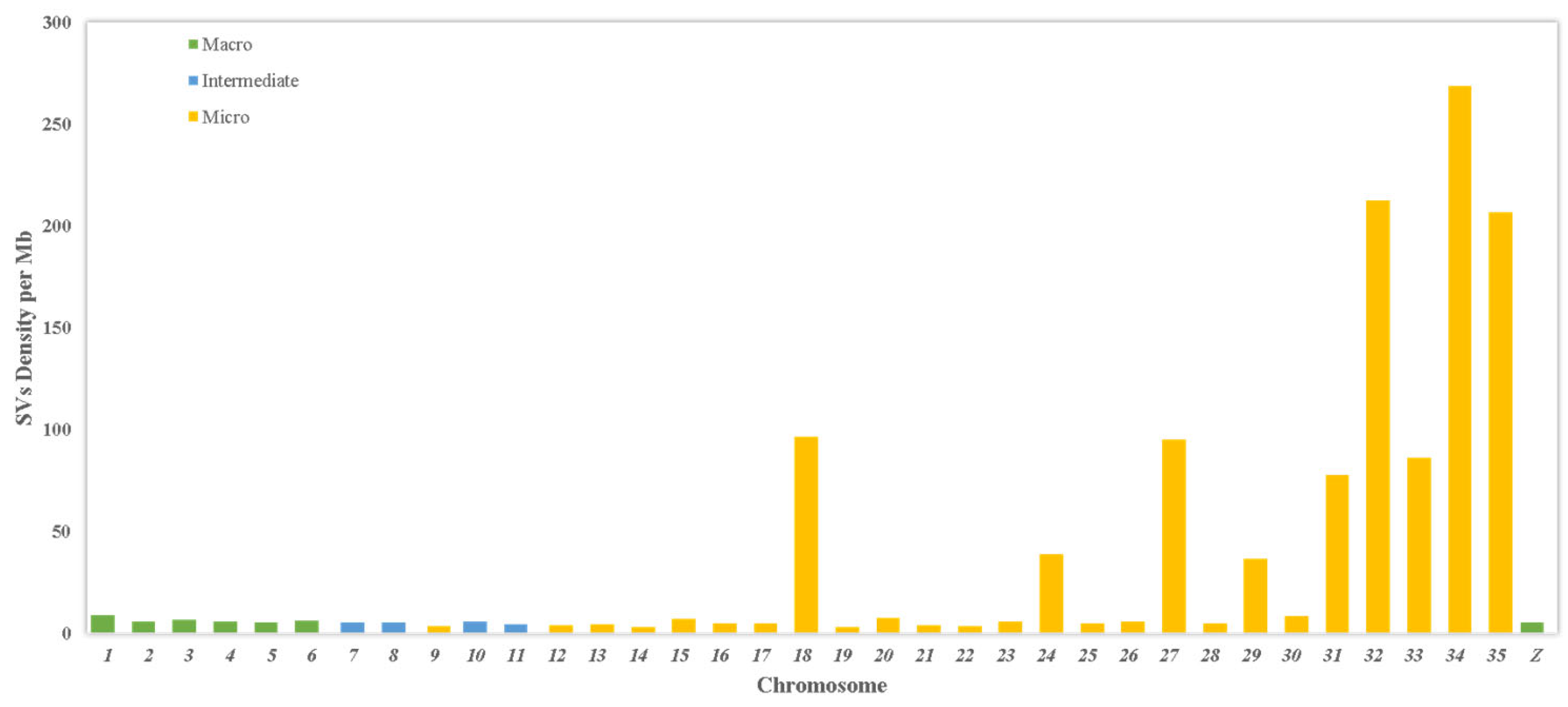
3.3. Distribution Across Chromosomes
3.4. Distribution of Structure Variations per Animal
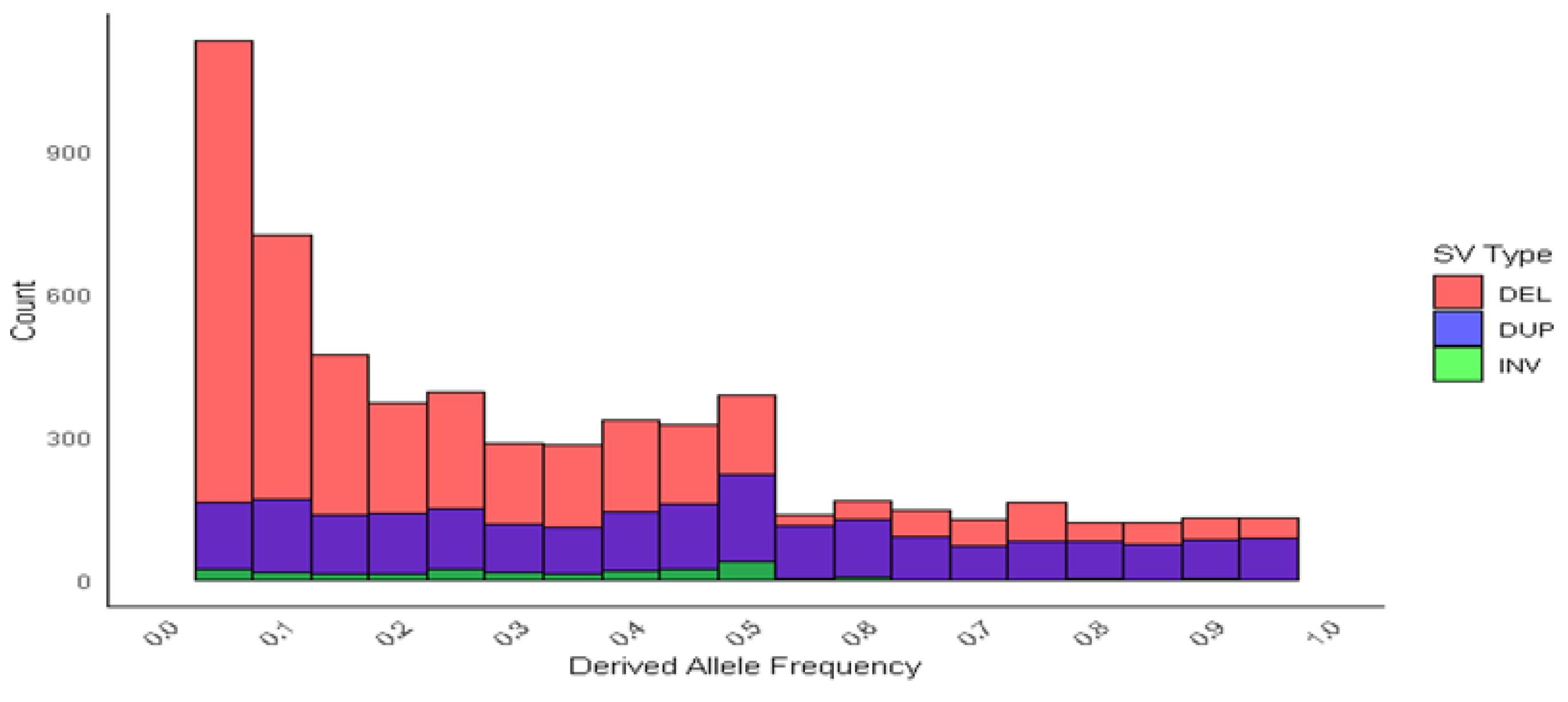
3.5. Allele Frequency
3.6. Functional Annotation
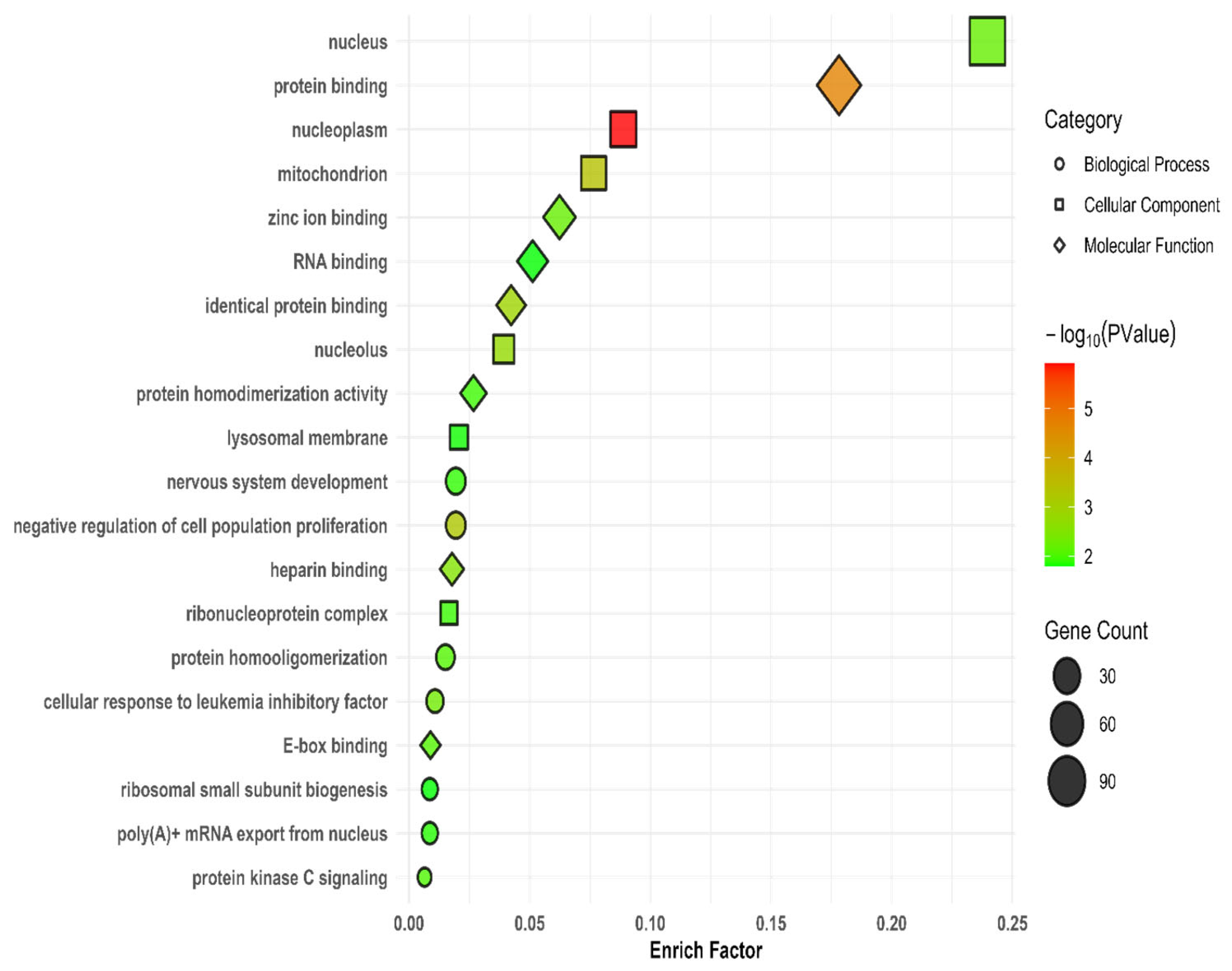
3.7. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baéza, E.; Guillier, L.; Petracci, M. Review: Production Factors Affecting Poultry Carcass and Meat Quality Attributes. Animal 2022, 16, 100331. [Google Scholar] [CrossRef] [PubMed]
- FAO FAOSTATA [Internet]. 2023. Available online: https://www.fao.org/faostat/en/#data/QCL/visualize (accessed on 22 June 2023).
- Thornton, E.K. Introduction to the Special Issue—Turkey Husbandry and Domestication: Recent Scientific Advances. J. Archaeol. Sci. Rep. 2016, 10, 514–519. [Google Scholar] [CrossRef]
- Speller, C.F.; Kemp, B.M.; Wyatt, S.D.; Monroe, C.; Lipe, W.D.; Arndt, U.M.; Yang, D.Y. Ancient Mitochondrial DNA Analysis Reveals Complexity of Indigenous North American Turkey Domestication. Proc. Natl. Acad. Sci. USA 2010, 107, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo, L.V.; Avellanet, R.; Azón, R.; Tejedor, M.T. Mitochondrial DNA Analysis in Two Heritage European Breeds Confirms Mesoamerican Origin and Low Genetic Variability of Domestic Turkey. Anim. Genet. 2013, 44, 786. [Google Scholar] [CrossRef] [PubMed]
- De Grossi Mazzorin, I.; Epifani, I. Introduzione e Diffusione in Italia Di Animali Esotici Dal Nuovo Mondo: Il Caso Del Tacchino (Meleagris gallopavo L.). L’Idomeneo 2016, 2015, 55–74. [Google Scholar]
- Maltin, E.; Jakobsson, H. The Introduction of the Turkey (Meleagris gallopavo) in Early Modern Sweden–Historical and Zooarchaeological Evidence of Husbandry and Consumption. Postmediev Archaeol. 2023, 57, 1–28. [Google Scholar] [CrossRef]
- Eiche, S. Presenting the Turkey: The Fabulous Story of a Flamboyant and Flavourful Bird; Centro Di: Florence, Italy, 2004; ISBN 8870384144. [Google Scholar]
- Bernini, F.; Bagnato, A.; Marelli, S.P.; Zaniboni, L.; Cerolini, S.; Strillacci, M.G. Genetic Diversity and Identification of Homozygosity-Rich Genomic Regions in Seven Italian Heritage Turkey (Meleagris gallopavo) Breeds. Genes 2021, 12, 1342. [Google Scholar] [CrossRef]
- Marelli, S.P.; Zaniboni, L.; Strillacci, M.G.; Madeddu, M.; Cerolini, S. Morphological Characterization of Two Light Italian Turkey Breeds. Animals 2022, 12, 571. [Google Scholar] [CrossRef]
- Marelli, S.P.; Colombo, E.; Mangiagalli, M.G.; Zaniboni, L.; Cassinelli, C.; Cerolini, S.; Guidobono Cavalchini, L. Phenotypical Characterization of Some Italian Turkey (Meleagris gallopavo) Breeds. 2009. Available online: https://www.semanticscholar.org/paper/Phenotypical-characterization-of-some-Italian-Marelli-Colombo/eb04b909f5d4b5bb2bbf51c8f5400a2ebf787030 (accessed on 1 January 2009).
- Colli, L.; Negrini, R.; Ajmone Marsan, P.; Consortium, G. Marcatori Molecolari, Genoma e Risorse Genetiche Animali. Fondazione Iniziative Zootecniche. La Salvaguardia della Biodiversita Animale. Iniziative Generali ed Azioni Intraprese in Italia a Tutela delle Razze Minacciate; Fondazione Iniziative Zooprofilattiche e Zootecniche: Brescia, Italy, 2011. [Google Scholar]
- Strillacci, M.G.; Gorla, E.; Ríos-Utrera, A.; Vega-Murillo, V.E.; Montaño-Bermudez, M.; Garcia-Ruiz, A.; Cerolini, S.; Román-Ponce, S.I.; Bagnato, A. Copy Number Variation Mapping and Genomic Variation of Autochthonous and Commercial Turkey Populations. Front. Genet. 2019, 10, 982. [Google Scholar] [CrossRef]
- Makanjuola, B.O.; Miglior, F.; Abdalla, E.A.; Maltecca, C.; Schenkel, F.S.; Baes, C.F. Effect of Genomic Selection on Rate of Inbreeding and Coancestry and Effective Population Size of Holstein and Jersey Cattle Populations. J. Dairy. Sci. 2020, 103, 5183–5199. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Bosse, M.; Takeda, H.; Moreira, G.C.M.; Karim, L.; Druet, T.; Oget-Ebrad, C.; Coppieters, W.; Veerkamp, R.F.; Groenen, M.A.M.; et al. High-Resolution Structural Variants Catalogue in a Large-Scale Whole Genome Sequenced Bovine Family Cohort Data. BMC Genom. 2023, 24, 225. [Google Scholar] [CrossRef] [PubMed]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome Structural Variation Discovery and Genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, R.E.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Hsi-Yang Fritz, M. An Integrated Map of Structural Variation in 2504 Human Genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef]
- Aslam, M.L.; Bastiaansen, J.W.M.; Megens, H.-J.; Crooijmans, R.P.M.A.; Nasreen, F.; Blomberg, L.A.; Van Tassell, C.P.; Sonstegard, T.S.; Schroeder, S.G.; Groenen, M.A.M.; et al. Genome-Wide Candidate Regions for Selective Sweeps Revealed through Massive Parallel Sequencing of DNA across Ten Turkey Populations. BMC Genet. 2014, 15, 117. [Google Scholar] [CrossRef]
- Strillacci, M.G.; Marelli, S.P.; Milanesi, R.; Zaniboni, L.; Punturiero, C.; Cerolini, S. Copy Number Variants in Four Italian Turkey Breeds. Animals 2021, 11, 391. [Google Scholar] [CrossRef]
- Fernandes, A.C.; da Silva, V.H.; Goes, C.P.; Moreira, G.C.M.; Godoy, T.F.; Ibelli, A.M.G.; Peixoto, J.D.O.; Cantão, M.E.; Ledur, M.C.; de Rezende, F.M. Genome-Wide Detection of CNVs and Their Association with Performance Traits in Broilers. BMC Genom. 2021, 22, 1–18. [Google Scholar] [CrossRef]
- Zhang, J.; Nie, C.; Li, X.; Zhao, X.; Jia, Y.; Han, J.; Chen, Y.; Wang, L.; Lv, X.; Yang, W. Comprehensive Analysis of Structural Variants in Chickens Using PacBio Sequencing. Front. Genet. 2022, 13, 971588. [Google Scholar] [CrossRef]
- Cendron, F.; Cassandro, M.; Penasa, M. Genome-Wide Investigation to Assess Copy Number Variants in the Italian Local Chicken Population. J. Anim. Sci. Biotechnol. 2024, 15, 2. [Google Scholar] [CrossRef]
- Li, R.; Gong, M.; Zhang, X.; Wang, F.; Liu, Z.; Zhang, L.; Yang, Q.; Xu, Y.; Xu, M.; Zhang, H. A Sheep Pangenome Reveals the Spectrum of Structural Variations and Their Effects on Tail Phenotypes. Genome Res. 2023, 33, 463–477. [Google Scholar] [CrossRef]
- Qiao, G.; Xu, P.; Guo, T.; He, X.; Yue, Y.; Yang, B. Genome-wide Detection of Structural Variation in Some Sheep Breeds Using Whole-genome Long-read Sequencing Data. J. Anim. Breed. Genet. 2024, 141, 403–414. [Google Scholar] [CrossRef]
- Boussaha, M.; Esquerré, D.; Barbieri, J.; Djari, A.; Pinton, A.; Letaief, R.; Salin, G.; Escudié, F.; Roulet, A.; Fritz, S. Genome-Wide Study of Structural Variants in Bovine Holstein, Montbéliarde and Normande Dairy Breeds. PLoS ONE 2015, 10, e0135931. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, M.; Derks, M.F.L.; Andersson, G.; Medugorac, I.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Introgression Contributes to Distribution of Structural Variations in Cattle. Genomics 2021, 113, 3092–3102. [Google Scholar] [CrossRef] [PubMed]
- Braga, L.G.; Chud, T.C.S.; Watanabe, R.N.; Savegnago, R.P.; Sena, T.M.; do Carmo, A.S.; Machado, M.A.; Panetto, J.C.d.C.; da Silva, M.V.G.B.; Munari, D.P. Identification of Copy Number Variations in the Genome of Dairy Gir Cattle. PLoS ONE 2023, 18, e0284085. [Google Scholar] [CrossRef]
- Zong, W.; Wang, J.; Zhao, R.; Niu, N.; Su, Y.; Hu, Z.; Liu, X.; Hou, X.; Wang, L.; Wang, L. Associations of Genome-Wide Structural Variations with Phenotypic Differences in Cross-Bred Eurasian Pigs. J. Anim. Sci. Biotechnol. 2023, 14, 136. [Google Scholar] [CrossRef]
- Yang, L.; Yin, H.; Bai, L.; Yao, W.; Tao, T.; Zhao, Q.; Gao, Y.; Teng, J.; Xu, Z.; Lin, Q. Mapping and Functional Characterization of Structural Variation in 1060 Pig Genomes. Genome Biol. 2024, 25, 116. [Google Scholar] [CrossRef]
- Saleh, M.S.; Landi, V.; Derks, M.F.L.; Centoducati, G.; Groenen, M.A.M.; De Palo, P.; Ciani, E.; Strillacci, M.G.; Bagnato, A.; Pugliese, N.; et al. Genome-Wide Analyses of Genomic Diversity, Population Structure and Selection Signatures in Italian Turkey Populations. Poult. Sci. 2025, 104, 104543. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Barros, C.P.; Derks, M.F.L.; Mohr, J.; Wood, B.J.; Crooijmans, R.P.M.A.; Megens, H.-J.; Bink, M.C.A.M.; Groenen, M.A.M. A New Haplotype-Resolved Turkey Genome to Enable Turkey Genetics and Genomics Research. Gigascience 2023, 12, giad051. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Subgroup, 1000 Genome Project Data Processing the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Chang, W.; Wickham, M.H. Package ‘Ggplot2.’ Create Elegant Data Visualisations Using the Grammar of Graphics, Version 2; MIT: Cambridge, MA, USA, 2016; pp. 1–189. [Google Scholar]
- Saleh, M.S.; Landi, V.; Derks, M.F.L.; Centoducati, G.; Groenen, M.A.M.; De Palo, P.; Ciani, E.; Pugliese, N.; Circella, E.; Camarda, A. Genomic Scans for Selection and Runs of Homozygosity in Southern Italian Turkey Populations. Poult. Sci. 2025, 104, 104750. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R Package for Biodiversity Analyses 2017. Available online: https://github.com/marcomilanesi/BITE (accessed on 29 August 2017).
- Layer, R.M.; Chiang, C.; Quinlan, A.R.; Hall, I.M. LUMPY: A Probabilistic Framework for Structural Variant Discovery. Genome Biol. 2014, 15, 1–19. [Google Scholar] [CrossRef]
- Chiang, C.; Layer, R.M.; Faust, G.G.; Lindberg, M.R.; Rose, D.B.; Garrison, E.P.; Marth, G.T.; Quinlan, A.R.; Hall, I.M. SpeedSeq: Ultra-Fast Personal Genome Analysis and Interpretation. Nat. Methods 2015, 12, 966–968. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Quinlan, A.R. Duphold: Scalable, Depth-Based Annotation and Curation of High-Confidence Structural Variant Calls. Gigascience 2019, 8, giz040. [Google Scholar] [CrossRef]
- Danecek, P.; McCarthy, S.A. BCFtools/Csq: Haplotype-Aware Variant Consequences. Bioinformatics 2017, 33, 2037–2039. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Bertolotti, A.C.; Layer, R.M.; Gundappa, M.K.; Gallagher, M.D.; Pehlivanoglu, E.; Nome, T.; Robledo, D.; Kent, M.P.; Røsæg, L.L.; Holen, M.M. The Structural Variation Landscape in 492 Atlantic Salmon Genomes. Nat. Commun. 2020, 11, 5176. [Google Scholar] [CrossRef]
- Turner, D.J.; Miretti, M.; Rajan, D.; Fiegler, H.; Carter, N.P.; Blayney, M.L.; Beck, S.; Hurles, M.E. Germline Rates of de Novo Meiotic Deletions and Duplications Causing Several Genomic Disorders. Nat. Genet. 2008, 40, 90–95. [Google Scholar] [CrossRef]
- Liao, Z.; Zhang, X.; Zhang, S.; Lin, Z.; Zhang, X.; Ming, R. Structural Variations in Papaya Genomes. BMC Genom. 2021, 22, 335. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Li, S.; Yao, W.; Xie, C.; Chen, Z.; Zeng, Z.; Wang, D.; Xu, K.; Shen, Z.; Mu, Y. The Meishan Pig Genome Reveals Structural Variation-mediated Gene Expression and Phenotypic Divergence Underlying Asian Pig Domestication. Mol. Ecol. Resour. 2021, 21, 2077–2092. [Google Scholar] [CrossRef] [PubMed]
- Kerstens, H.H.D.; Crooijmans, R.P.M.A.; Dibbits, B.W.; Vereijken, A.; Okimoto, R.; Groenen, M.A.M. Structural Variation in the Chicken Genome Identified by Paired-End Next-Generation DNA Sequencing of Reduced Representation Libraries. BMC Genom. 2011, 12, 94. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length/Type | Deletions | Duplications | Inversions |
|---|---|---|---|
| 0–50 bp | 274 | 0 | 13 |
| 50–100 bp | 1062 | 0 | 11 |
| 100–1000 bp | 3237 | 1028 | 142 |
| 1000–10,000 bp | 1581 | 984 | 273 |
| >10,000 bp | 558 | 659 | 991 |
| Total | 6712 | 2671 | 1430 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saleh, M.S.; Landi, V.; Derks, M.F.L.; Centoducati, G.; Groenen, M.A.M.; De Palo, P.; Ciani, E.; Pugliese, N.; Circella, E.; Camarda, A. Genome Wide Identification of Structure Variations in Five Italian Turkey Populations. Animals 2025, 15, 339. https://doi.org/10.3390/ani15030339
Saleh MS, Landi V, Derks MFL, Centoducati G, Groenen MAM, De Palo P, Ciani E, Pugliese N, Circella E, Camarda A. Genome Wide Identification of Structure Variations in Five Italian Turkey Populations. Animals. 2025; 15(3):339. https://doi.org/10.3390/ani15030339
Chicago/Turabian StyleSaleh, Medhat S., Vincenzo Landi, Martijn F. L. Derks, Gerardo Centoducati, Martien A. M. Groenen, Pasquale De Palo, Elena Ciani, Nicola Pugliese, Elena Circella, and Antonio Camarda. 2025. "Genome Wide Identification of Structure Variations in Five Italian Turkey Populations" Animals 15, no. 3: 339. https://doi.org/10.3390/ani15030339
APA StyleSaleh, M. S., Landi, V., Derks, M. F. L., Centoducati, G., Groenen, M. A. M., De Palo, P., Ciani, E., Pugliese, N., Circella, E., & Camarda, A. (2025). Genome Wide Identification of Structure Variations in Five Italian Turkey Populations. Animals, 15(3), 339. https://doi.org/10.3390/ani15030339