Influence of Pregnancy on Whole-Transcriptome Sequencing in the Mammary Gland of Kazakh Mares

 , , and
, , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animal
2.2. Transcriptome Sequencing
2.3. Data Quality Control
2.4. Relationship Analysis of Samples
2.5. Differential Expression Analysis
2.6. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
2.7. RT-qPCR Validation
2.8. Statistical Analysis
3. Results
3.1. Expression of mRNAs, lncRNAs, miRNAs, and circRNAs in the Samples
3.2. Correlation and Clustering Analysis of mRNAs, lncRNAs, miRNAs, and circRNAs
3.3. PCA of mRNAs, lncRNAs, miRNAs, and circRNAs
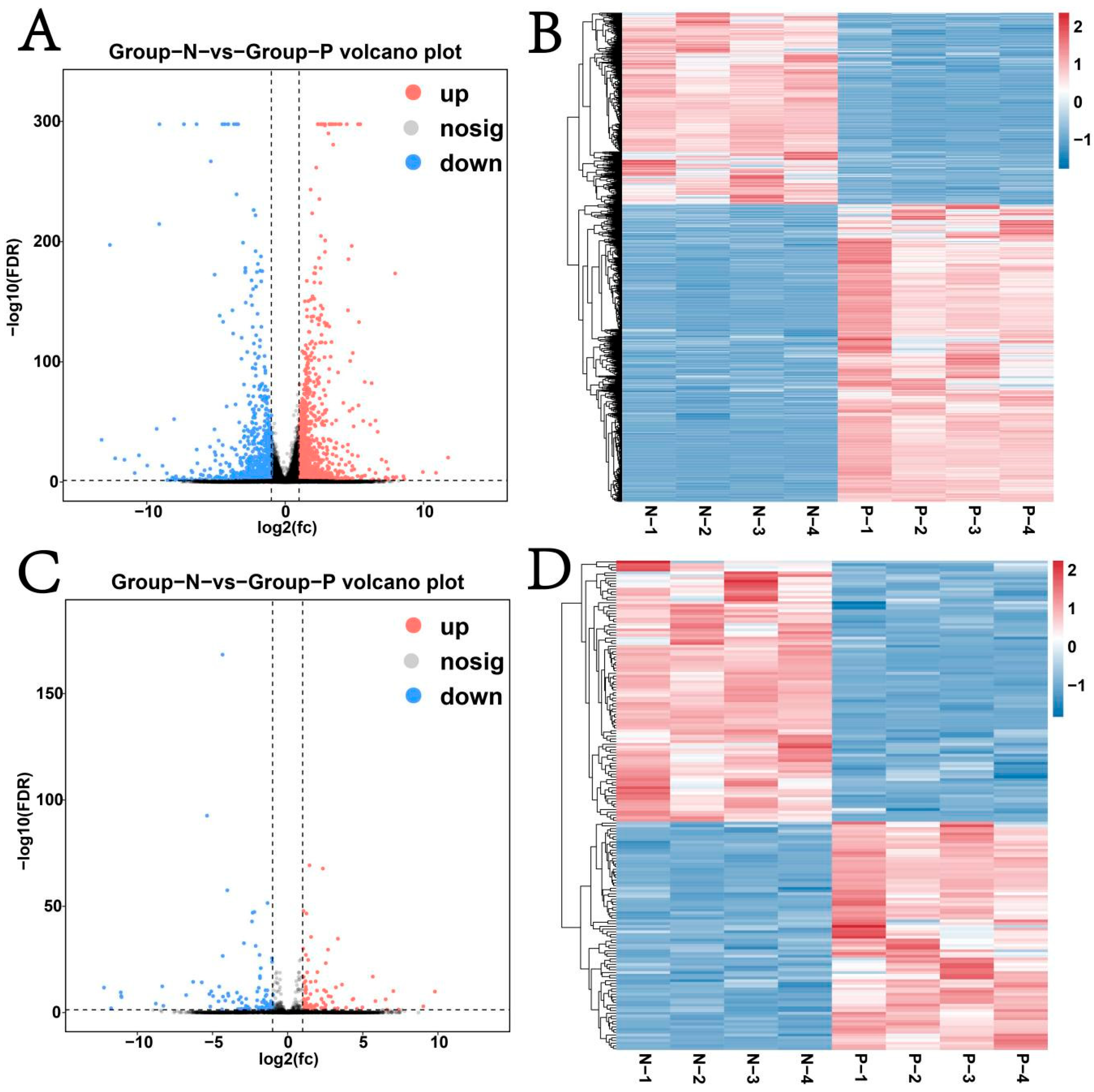
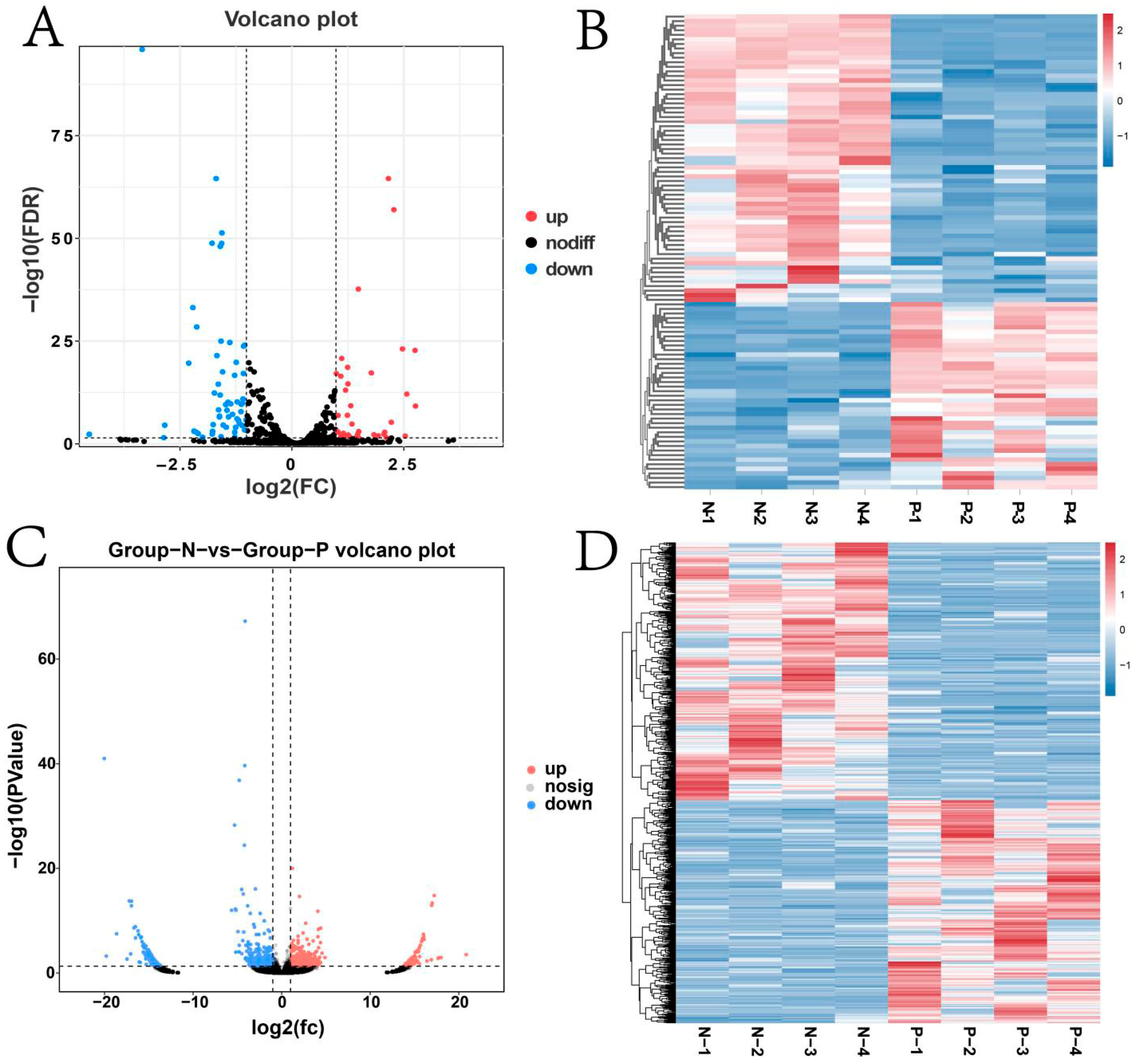
3.4. Analysis of Differential Expression of mRNAs, lncRNAs, miRNAs, and circRNAs
3.5. GO and KEGG Enrichment Analysis
3.6. Gene Interaction Network Analysis
3.7. Association Analyses of mRNAs and lncRNAs
3.8. Cis-Regulatory Elements Analysis
3.9. Trans-Regulatory Elements Analysis
3.10. ceRNA Analysis
3.11. RT-qPCR Analysis
4. Discussion
4.1. Differential RNA Analysis of the Mammary Gland Across Pregnant and Non-Pregnant Kazakh Mares
4.2. Participation of Cytokine–Cytokine Receptor Interaction in Lactation Stimulation
4.3. Function of the ceRNA Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ren, W.; Wang, J.; Zeng, Y.; Wang, T.; Sun, Z.; Meng, J.; Yao, X. Investigating age-related differences in muscles of Kazakh horse through transcriptome analysis. Gene 2024, 919, 148483. [Google Scholar] [CrossRef] [PubMed]
- Pozharskiy, A.; Abdrakhmanova, A.; Beishova, I.; Shamshidin, A.; Nametov, A.; Ulyanova, T.; Bekova, G.; Kikebayev, N.; Kovalchuk, A.; Ulyanov, V. Genetic structure and genome-wide association study of the traditional Kazakh horses. Animal 2023, 17, 100926. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Y.; Ma, H.; Cao, H.; Liu, W. Integrating genome-wide methylation and transcriptome-wide analyses to reveal the genetic mechanism of milk traits in Kazakh horses. Gene 2023, 856, 147143. [Google Scholar] [CrossRef] [PubMed]
- Ulaangerel, T.; Wang, M.; Zhao, B.; Yi, M.; Shen, Y.; Mengkh, Y.; Wen, X.; Dugarjav, M.; Bou, G. A Comparative Analysis of the Gene Expression Profiles in the Mammary Glands of Lactating and Nonlactating Mares at the Second Month of Gestation. Animals 2024, 14, 2319. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Marins, T.; Calix, J.; Qi, Z.; Savegnago, C.; Roper, A.; Woldemeskel, M.; Bernard, J.; Tao, S. Systemic and mammary inflammation and mammary gland development of Holstein dairy cows around dry-off and calving. J. Dairy Sci. 2025, 108, 2090–2110. [Google Scholar] [CrossRef]
- Hughes, K. Development and pathology of the equine mammary gland. J. Mammary Gland. Biol. Neoplasia 2021, 26, 121–134. [Google Scholar] [CrossRef]
- Anantamongkol, U.; Charoenphandhu, N.; Wongdee, K.; Teerapornpuntakit, J.; Suthiphongchai, T.; Prapong, S.; Krishnamra, N. Transcriptome analysis of mammary tissues reveals complex patterns of transporter gene expression during pregnancy and lactation. Cell Biol. Int. 2010, 34, 67–74. [Google Scholar] [CrossRef]
- Qi, Y.-X.; Liu, Y.-B.; Rong, W.-H. RNA-Seq and its applications: A new technology for transcriptomics. Hereditas 2011, 33, 1191–1202. [Google Scholar] [CrossRef]
- Hosseinzadeh, S.; Rafat, S.A.; Javanmard, A.; Fang, L. Identification of candidate genes associated with milk production and mastitis based on transcriptome-wide association study. Anim. Genet. 2024, 55, 430–439. [Google Scholar] [CrossRef]
- Xia, W.; Liu, Y.; Loor, J.J.; Bionaz, M.; Jiang, M. Dynamic profile of the yak mammary transcriptome during the lactation cycle. Animals 2023, 13, 1710. [Google Scholar] [CrossRef]
- Shi, T.P.; Zhang, L. Application of whole transcriptomics in animal husbandry. Hereditas 2019, 41, 193–205. [Google Scholar] [PubMed]
- Ren, W.; Wang, J.; Zeng, Y.; Wang, T.; Meng, J.; Yao, X. Transcriptome identification of differential mammary genes of Kazakh horses during early pregnancy. Gene 2024, 902, 148189. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.J.; Moorehead, R.A. The miR-200 family in normal mammary gland development. BMC Dev. Biol. 2021, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Zhu, Y. Exploration of Key Regulators of Back Fat Deposition in Dahe Pigs Based on Whole Transcriptome Sequencing; Yunnan Agricultural University: Kunming, China, 2024. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Spina, E.; Cowin, P. Embryonic mammary gland development. In Seminars in Cell & Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 114, pp. 83–92. [Google Scholar]
- Wang, W.; Wang, S.; Wang, H.; Zheng, E.; Wu, Z.; Li, Z. Protein Dynamic Landscape during Mouse Mammary Gland Development from Virgin to Pregnant, Lactating, and Involuting Stages. J. Agric. Food Chem. 2024, 72, 7546–7557. [Google Scholar] [CrossRef]
- McNally, S.; Stein, T. Overview of mammary gland development: A comparison of mouse and human. In Mammary Gland Development: Methods and Protocols; Martin, F., Stein, T., Howlin, J., Eds.; Humana Press: New York, NY, USA, 2017; pp. 1–17. [Google Scholar]
- Wang, W.; Zang, X.; Liu, Y.; Liang, Y.; Cai, G.; Wu, Z.; Li, Z. Dynamic miRNA landscape links mammary gland development to the regulation of milk protein expression in mice. Animals 2022, 12, 727. [Google Scholar] [CrossRef]
- Jena, M.K.; Jaswal, S.; Kumar, S.; Mohanty, A.K. Molecular mechanism of mammary gland involution: An update. Dev. Biol. 2019, 445, 145–155. [Google Scholar] [CrossRef]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef]
- Alateyah, N.; Gupta, I.; Rusyniak, R.S.; Ouhtit, A. SOD2, a potential transcriptional target underpinning CD44-promoted breast cancer progression. Molecules 2022, 27, 811. [Google Scholar] [CrossRef]
- Sun, L.; Wang, F.; Wu, Z.; Ma, L.; Baumrucker, C.; Bu, D. Comparison of selenium source in preventing oxidative stress in bovine mammary epithelial cells. Animals 2020, 10, 842. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, J.-Y.; Yu, Q.; Jin, J.-O. Interleukin-7 produced by intestinal epithelial cells in response to Citrobacter rodentium infection plays a major role in innate immunity against this pathogen. Infect. Immun. 2015, 83, 3213–3223. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Lai, Y.T.; Rahman, M.M.; Chen, H.W.; Husna, A.A.; Fujikawa, T.; Ando, T.; Kitahara, G.; Koiwa, M.; Kubota, C. Bovine milk transcriptome analysis reveals microRNAs and RNU2 involved in mastitis. FEBS J. 2020, 287, 1899–1918. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Wu, J.; Han, P.; Zhao, Z.; Song, X. Circular RNA expression profiles and features in human tissues: A study using RNA-seq data. BMC Genom. 2017, 18, 131–142. [Google Scholar] [CrossRef]
- Wang, J.; Ke, N.; Wu, X.; Zhen, H.; Hu, J.; Liu, X.; Li, S.; Zhao, F.; Li, M.; Shi, B. MicroRNA-148a Targets DNMT1 and PPARGC1A to Regulate the Viability, Proliferation, and Milk Fat Synthesis of Ovine Mammary Epithelial Cells. Int. J. Mol. Sci. 2024, 25, 8558. [Google Scholar] [CrossRef]
- Wang, X.-P.; Luoreng, Z.-M.; Zan, L.-S.; Li, F.; Li, N. Bovine miR-146a regulates inflammatory cytokines of bovine mammary epithelial cells via targeting the TRAF6 gene. J. Dairy Sci. 2017, 100, 7648–7658. [Google Scholar] [CrossRef]
- Watson, C.J.; Oliver, C.H.; Khaled, W.T. Cytokine signalling in mammary gland development. J. Reprod. Immunol. 2011, 88, 124–129. [Google Scholar] [CrossRef]
- Spangler, J.B.; Moraga, I.; Mendoza, J.L.; Garcia, K.C. Insights into cytokine–receptor interactions from cytokine engineering. Annu. Rev. Immunol. 2015, 33, 139–167. [Google Scholar] [CrossRef]
- Sun, X.; Ingman, W.V. Cytokine networks that mediate epithelial cell-macrophage crosstalk in the mammary gland: Implications for development and cancer. J. Mammary Gland. Biol. Neoplasia 2014, 19, 191–201. [Google Scholar] [CrossRef]
- Zhao, W.; Shahzad, K.; Jiang, M.; Graugnard, D.E.; Rodriguez-Zas, S.L.; Luo, J.; Loor, J.J.; Hurley, W.L. Bioinformatics and gene network analyses of the swine mammary gland transcriptome during late gestation. Bioinform. Biol. Insights 2013, 7, S12205. [Google Scholar] [CrossRef]
- Alluwaimi, A.M. The cytokines of bovine mammary gland: Prospects for diagnosis and therapy. Res. Vet. Sci. 2004, 77, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Jiang, Y.; Zhu, J.; He, Y.; Yin, H.; Duan, Q.; Zhang, L.; Cao, B.; An, X. CircRNA8220 sponges MiR-8516 to regulate cell viability and milk synthesis via Ras/MEK/ERK and PI3K/AKT/mTOR pathways in goat mammary epithelial cells. Animals 2020, 10, 1347. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, L.M.; Piek, E.; Böttinger, E.P. TGF-β signaling in mammary gland development and tumorigenesis. J. Mammary Gland. Biol. Neoplasia 2001, 6, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Brisken, C.; Ayyannan, A.; Nguyen, C.; Heineman, A.; Reinhardt, F.; Jan, T.; Dey, S.; Dotto, G.P.; Weinberg, R.A. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev. Cell 2002, 3, 877–887. [Google Scholar] [CrossRef]
- Mu, T.; Hu, H.; Ma, Y.; Yang, C.; Feng, X.; Wang, Y.; Liu, J.; Yu, B.; Zhang, J.; Gu, Y. Identification of critical lncRNAs for milk fat metabolism in dairy cows using WGCNA and the construction of a ceRNAs network. Anim. Genet. 2022, 53, 740–760. [Google Scholar] [CrossRef]
- Chen, M.; Wei, X.; Song, M.; Jiang, R.; Huang, K.; Deng, Y.; Liu, Q.; Shi, D.; Li, H. Circular RNA circMYBPC1 promotes skeletal muscle differentiation by targeting MyHC. Mol. Ther.-Nucleic Acids 2021, 24, 352–368. [Google Scholar] [CrossRef]
- Lawless, N.; Vegh, P.; O’Farrelly, C.; Lynn, D.J. The role of microRNAs in bovine infection and immunity. Front. Immunol. 2014, 5, 611. [Google Scholar] [CrossRef]
- Bach, K.; Pensa, S.; Grzelak, M.; Hadfield, J.; Adams, D.J.; Marioni, J.C.; Khaled, W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017, 8, 2128. [Google Scholar] [CrossRef]
- Ostrander, E.A.; Wang, G.-D.; Larson, G.; Vonholdt, B.M.; Davis, B.W.; Jagannathan, V.; Hitte, C.; Wayne, R.K.; Zhang, Y.-P. Dog10K: An international sequencing effort to advance studies of canine domestication, phenotypes and health. Natl. Sci. Rev. 2019, 6, 810–824. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA | KEGG_A_Class | KEGG_B_Class | Pathway | Group-N-vs-Group-P | All | p Value | Q Value | Pathway ID |
|---|---|---|---|---|---|---|---|---|
| mRNAs | Metabolism | Global and overview maps | Metabolic pathways | 145 | 1516 | 0.995437 | 1.000000 × 10+0 | ko01100 |
| Human Diseases | Cancer: Overview | Pathways in cancer | 101 | 537 | 1.630413 × 10−7 | 3.586909 × 10−6 | ko05200 | |
| Environmental Information Processing | Signaling molecules and interaction | Cytokine–cytokine receptor interaction | 95 | 291 | 7.933928 × 10−23 | 2.618196 × 10−20 | ko04060 | |
| miRNAs | Metabolism | Global and overview maps | Metabolic pathways | 1287 | 3551 | 0.00065877 | 3.238181 × 10−3 | ko01100 |
| Human Diseases | Cancer: Overview | Pathways in cancer | 527 | 1334 | 5.577158 × 10−6 | 5.406745 × 10−5 | ko05200 | |
| Environmental Information Processing | Signal transduction | PI3K-Akt signaling pathway | 381 | 916 | 4.260542 × 10−7 | 5.718958 × 10−6 | ko04151 | |
| circRNAs | Metabolism | Global and overview maps | Metabolic pathways | 72 | 1513 | 0.09367813 | 0.61050942 | ko01100 |
| Human Diseases | Infectious disease: bacterial | Pathogenic Escherichia coli infection | 21 | 241 | 0.000910662 | 0.08811273 | ko05130 | |
| Cellular Processes | Transport and catabolism | Endocytosis | 20 | 261 | 0.005352532 | 0.18711521 | ko04144 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Lu, Z.; Yao, X.; Li, L.; Meng, J.; Wang, J.; Zeng, Y.; Ren, W. Influence of Pregnancy on Whole-Transcriptome Sequencing in the Mammary Gland of Kazakh Mares. Animals 2025, 15, 2056. https://doi.org/10.3390/ani15142056
Zhang Z, Lu Z, Yao X, Li L, Meng J, Wang J, Zeng Y, Ren W. Influence of Pregnancy on Whole-Transcriptome Sequencing in the Mammary Gland of Kazakh Mares. Animals. 2025; 15(14):2056. https://doi.org/10.3390/ani15142056
Chicago/Turabian StyleZhang, Zhenyu, Zhixin Lu, Xinkui Yao, Linling Li, Jun Meng, Jianwen Wang, Yaqi Zeng, and Wanlu Ren. 2025. "Influence of Pregnancy on Whole-Transcriptome Sequencing in the Mammary Gland of Kazakh Mares" Animals 15, no. 14: 2056. https://doi.org/10.3390/ani15142056
APA StyleZhang, Z., Lu, Z., Yao, X., Li, L., Meng, J., Wang, J., Zeng, Y., & Ren, W. (2025). Influence of Pregnancy on Whole-Transcriptome Sequencing in the Mammary Gland of Kazakh Mares. Animals, 15(14), 2056. https://doi.org/10.3390/ani15142056