
Impacts of Captive Domestication and Geographical Divergence on the Gut Microbiome of Endangered Forest Musk Deer
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Morphological and Molecular Biological Identification of Samples
2.2.1. Fecal Morphological Analysis
2.2.2. Molecular Biological Identification: DNA Extraction and PCR Amplification
- MBshortF4: 5′-TAGGTTAAATAGACCAAGAGCCTTCA-3′;
- MBshortR4: 5′-AGTTCGGCACGGATTAGCAG-3′.
2.3. DNA Extraction and Quality Control
2.4. Library Preparation and Sequencing
2.5. Bioinformatic Analysis
2.5.1. Data Preprocessing
2.5.2. Metagenomic Assembly and Gene Catalog Construction
2.5.3. Taxonomic Profiling and Functional
3. Results
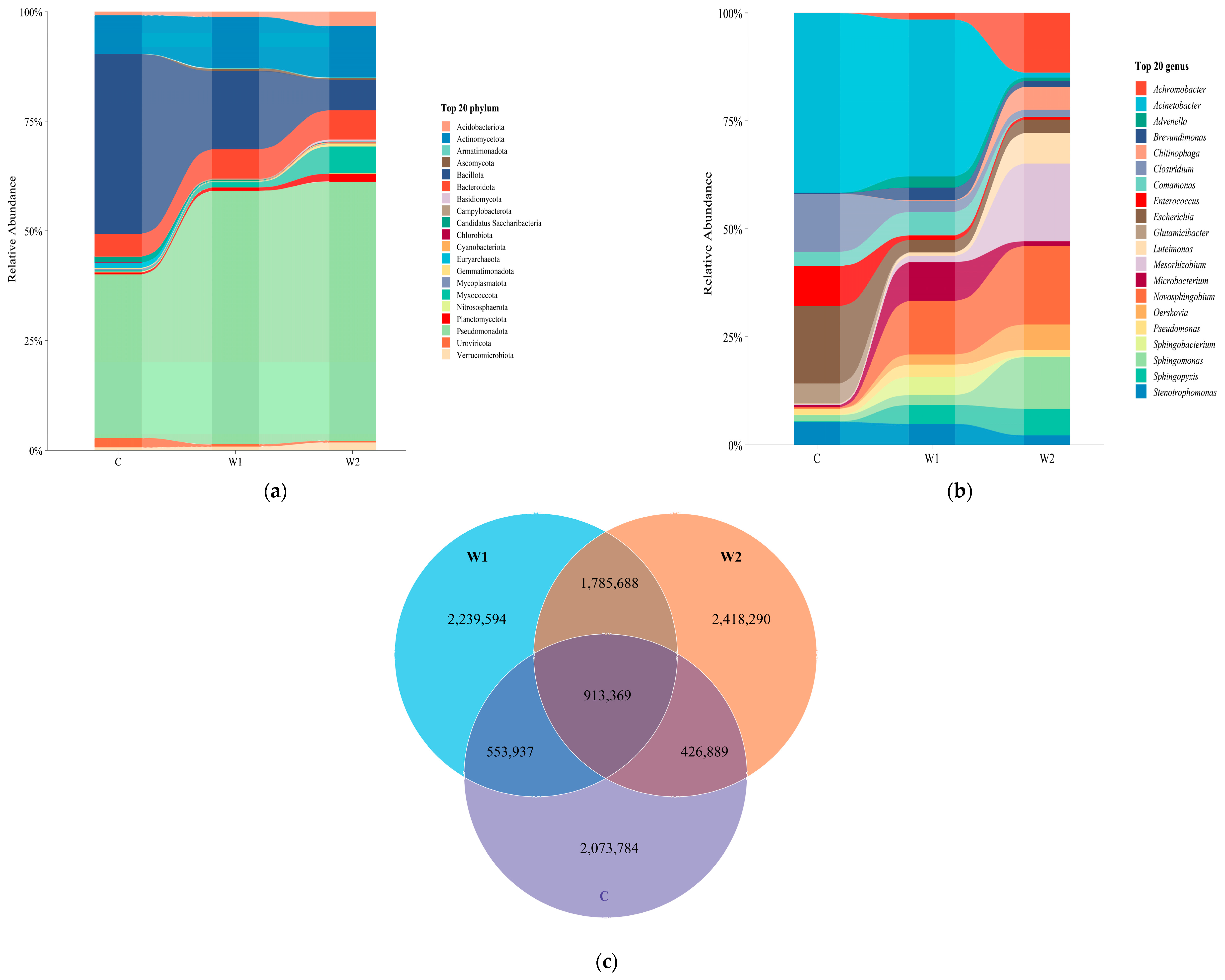
3.1. Gut Microbial Community Composition
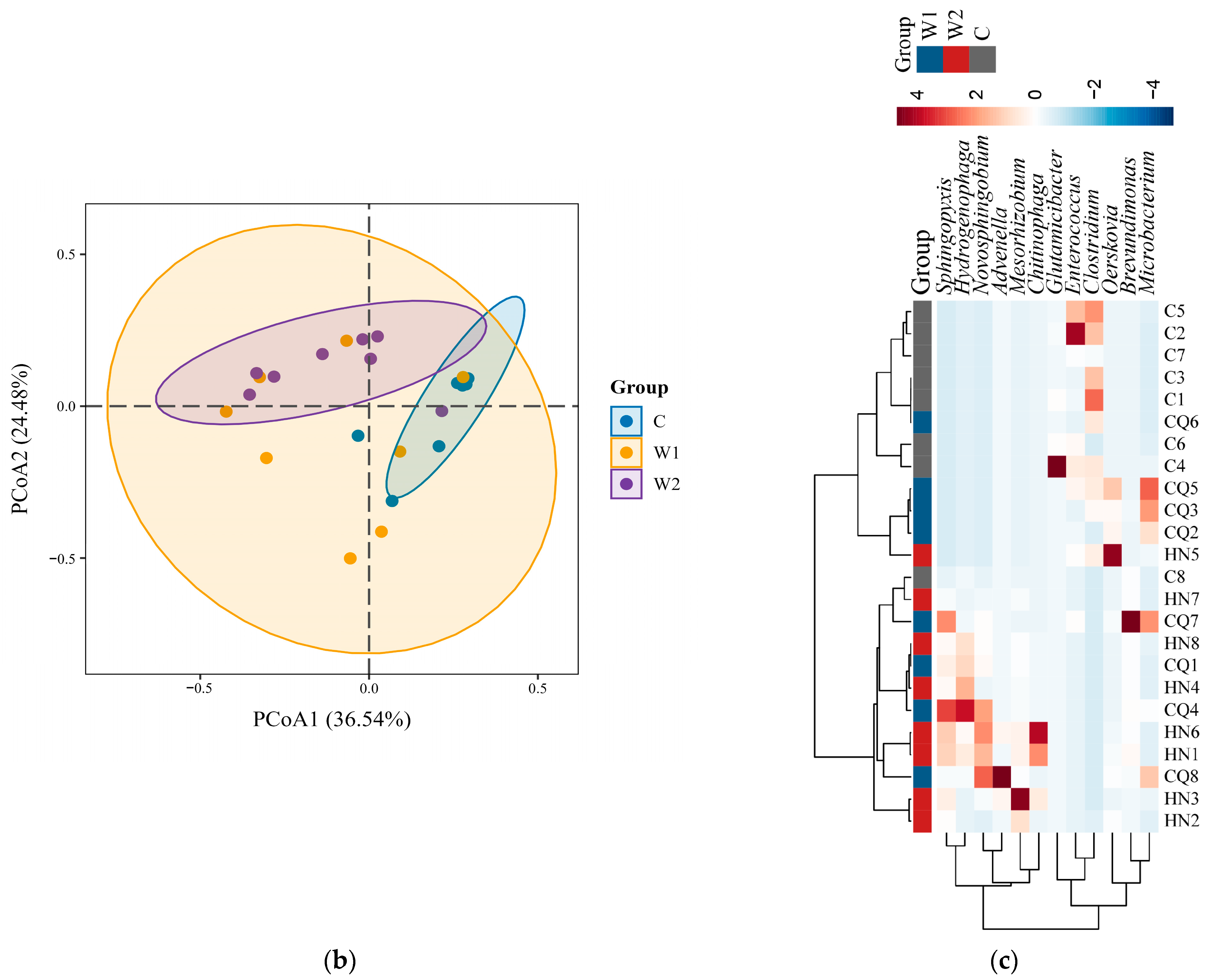
3.2. Comparative Analysis of Gut Microbial Diversity and Differences
3.3. Functional Composition of Gut Microbiota
4. Discussion
5. Conclusions
- (1)
- Optimizing captive environments by simulating natural habitat vegetation and dietary diversity to mitigate declines in gut microbial diversity;
- (2)
- Enhancing monitoring of pathogenic bacteria in captive individuals to develop scientific health management protocols;
- (3)
- Promoting data sharing and collaborative research across geographic regions to better understand and protect the gut microbial communities of this endangered species.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Aquila, P.; Lynn Carelli, L.; De Rango, F.; Passarino, G.; Bellizzi, D. Gut Microbiota as Important Mediator Between Diet and DNA Methylation and Histone Modifications in the Host. Nutrients 2020, 12, 597. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current Understanding of the Human Microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Panigrahi, P. The Neonatal Gut Microbiome and Global Health. Gut Microbes 2024, 16, 2352175. [Google Scholar] [CrossRef] [PubMed]
- West, A.G.; Waite, D.W.; Deines, P.; Bourne, D.G.; Digby, A.; McKenzie, V.J.; Taylor, M.W. The Microbiome in Threatened Species Conservation. Biol. Conserv. 2019, 229, 85–98. [Google Scholar] [CrossRef]
- Dearing, M.D.; Weinstein, S.B. Metabolic Enabling and Detoxification by Mammalian Gut Microbes. Annu. Rev. Microbiol. 2022, 76, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Arroyo-Rodríguez, V.; Dias, P.A.D. Effects of Habitat Fragmentation and Disturbance on Howler Monkeys: A Review. Am. J. Primatol. 2010, 72, 1–16. [Google Scholar] [CrossRef]
- Fogel, A.T. The Gut Microbiome of Wild Lemurs: A Comparison of Sympatric Lemur Catta and Propithecus Verreauxi. Folia Primatol. 2015, 86, 85–95. [Google Scholar] [CrossRef]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Rex Gaskins, H.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat Degradation Impacts Black Howler Monkey (Alouatta pigra) Gastrointestinal Microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R.; et al. Evolution of Mammals and Their Gut Microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Guo, W.; Mishra, S.; Wang, C.; Zhang, H.; Ning, R.; Kong, F.; Zeng, B.; Zhao, J.; Li, Y. Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca). Genes 2019, 10, 827. [Google Scholar] [CrossRef]
- Tang, L.; Li, Y.; Srivathsan, A.; Gao, Y.; Li, K.; Hu, D.; Zhang, D. Gut Microbiomes of Endangered Przewalski’s Horse Populations in Short- and Long-Term Captivity: Implication for Species Reintroduction Based on the Soft-Release Strategy. Front. Microbiol. 2020, 11, 363. [Google Scholar] [CrossRef]
- San Juan, P.A.; Castro, I.; Dhami, M.K. Captivity Reduces Diversity and Shifts Composition of the Brown Kiwi Microbiome. Anim. Microbiome 2021, 3, 48. [Google Scholar] [CrossRef]
- Bueno de Mesquita, C.P.; Nichols, L.M.; Gebert, M.J.; Vanderburgh, C.; Bocksberger, G.; Lester, J.D.; Kalan, A.K.; Dieguez, P.; McCarthy, M.S.; Agbor, A.; et al. Structure of Chimpanzee Gut Microbiomes across Tropical Africa. mSystems 2021, 6, e01269-20. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Wang, B.; Tan, J.; Zhu, L.; Lou, D.; Cen, X. Comparative Analysis of the Gut Microbiota of Black Bears in China Using High-Throughput Sequencing. Mol. Genet. Genomics 2017, 292, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yuan, X.; Zhou, H.; Zhang, C.; Peng, J.; Peng, J.; Deng, Y.; Huang, P.; Gao, H.; Sun, N. Moschus berezovskii in Chongqing Jinfo Mountain National Nature Reserve:Population Size and Geographical Distribution. Chin. Agric. Sci. Bull. 2020, 36, 44–47. [Google Scholar]
- Zhao, J.; Wang, Q.; Zheng, C.; Hu, J.; Wang, R.; Jiang, G. Research on the suitable habitat for Moschus berezovskii based on maximum entropy model (MaxEnt) in climatic background. Hubei Agric. Sci. 2023, 62, 218–223. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, M.; Chang, F.; Wang, J.; Wang, Y.; Tang, J.; Zhang, K.; Gao, L.; Xue, X.; Wan, Y. The Essential Differences in Microbial and Chemical Components of Musk of Different Qualities Secreted by Captive Male Forest Musk Deer (Moschus berezovskii). Microb. Biotechnol. 2022, 15, 1783–1794. [Google Scholar] [CrossRef]
- Liu, C.; Hong, T.; Yu, L.; Chen, Y.; Dong, X.; Ren, Z. Single-Nucleus Multiomics Unravels the Genetic Mechanisms Underlying Musk Secretion in Chinese Forest Musk Deer (Moschus berezovskii). Int. J. Biol. Macromol. 2024, 279, 135050. [Google Scholar] [CrossRef]
- Xu, K.; Pu, S.; Liang, Z.; Wang, H.; Luo, C.; Zhu, C. Research progress in forest musk deer. Heilongjiang Anim. Sci. Vet. 2014, 147–150. [Google Scholar] [CrossRef]
- Yang, J.; Peng, G.; Shu, F.; Dong, D.; Zheng, X.; Zhu, C.; Li, X.; Ma, J.; Pan, C.; Yang, F.; et al. Characteristics of Steroidogenesis-Related Factors in the Musk Gland of Chinese Forest Musk Deer (Moschus berezovskii). J. Steroid Biochem. Mol. Biol. 2021, 212, 105916. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, B.; Chang, J.; Hu, X.; Li, C.; Xu, T.; Liu, S.; Hu, D. Population Genomics Reveals Moderate Genetic Differentiation between Populations of Endangered Forest Musk Deer Located in Shaanxi and Sichuan. BMC Genom. 2022, 23, 668. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, W.; Cheng, J.; Luo, Y.; Fu, W.; Zhou, L.; Wu, J.; Wang, Y.; Zhong, Z.; Yang, Z.; et al. Molecular Cloning, Prokaryotic Expression and Its Application Potential Evaluation of Interferon (IFN)-ω of Forest Musk Deer. Sci. Rep. 2023, 13, 10625. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Yang, S.; Zhou, J.; Zhang, T.; Qi, L.; Sun, X.; Fan, M.; Xu, S.; Cha, M.; et al. Comparative Analysis of the Gut Microbiota Composition between Captive and Wild Forest Musk Deer. Front. Microbiol. 2017, 8, 1705. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic Atherosclerosis Is Associated with an Altered Gut Metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of Intestinal Prevotella Copri Correlates with Enhanced Susceptibility to Arthritis. eLlife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and Assembly of Genomes and Genetic Elements in Complex Metagenomic Samples without Using Reference Genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A Human Gut Microbial Gene Catalogue Established by Metagenomic Sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary Intervention Impact on Gut Microbial Gene Richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative Analysis of Environmental Sequences Using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. An Integrated Catalog of Reference Genes in the Human Gut Microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Cao, X.; Zheng, X.; Wang, D.; Wang, H.; Jiang, B.; Bu, S. Composition of Microbiota and Antibiotic Resistance Genes in Rumen, Small Intestine and Large Intestine of Wild Forest Musk Deer Analyzed by Metagenomic Sequencing. Chin. J. Anim. Nutr. 2021, 33, 484–493. [Google Scholar] [CrossRef]
- Du, M.; Liang, Z.; Zhang, J.; Ding, X. Recent Advances on Interaction and Regulation between Intestinal Microflora and Host Mucosal Immune System of Ruminants. Chin. J. Anim. Nutr. 2021, 33, 2483–2494. [Google Scholar]
- Xie, F.; Jin, W.; Si, H.; Yuan, Y.; Tao, Y.; Liu, J.; Wang, X.; Yang, C.; Li, Q.; Yan, X.; et al. An Integrated Gene Catalog and over 10,000 Metagenome-Assembled Genomes from the Gastrointestinal Microbiome of Ruminants. Microbiome 2021, 9, 137. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, L.; Yan, T.; Yang, Y.; Xu, Q.; Wang, J.; Guo, W. Exploring the diversity of musk deer fecal bacterial community. J. China Agric. Univ. 2016, 21, 100–106. [Google Scholar] [CrossRef]
- Li, Q.S.; Wang, R.; Ma, Z.Y.; Zhang, X.M.; Jiao, J.Z.; Zhang, Z.G.; Ungerfeld, E.M.; Yi, K.L.; Zhang, B.Z.; Long, L.; et al. Dietary Selection of Metabolically Distinct Microorganisms Drives Hydrogen Metabolism in Ruminants. ISME J. 2022, 16, 2535–2546. [Google Scholar] [CrossRef]
- Chen, W.; Chen, X.; Zhang, Y.; Wu, H.; Zhao, D. Variation on Gut Microbiota Diversity of Endangered Red Pandas (Ailurus Fulgens) Living in Captivity Acrosss Geographical Latitudes. Front. Microbiol. 2024, 15, 1420305. [Google Scholar] [CrossRef]
- Bolam, D.N.; Sonnenburg, J.L. Mechanistic Insight into Polysaccharide Use within the Intestinal Microbiota. Gut Microbes 2011, 2, 86–90. [Google Scholar] [CrossRef]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the Gut Microbiota in Disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, Y.; Zhao, L.; Lin, S.; Luan, Q. Screening and Identification of Lignin-Degrading Bacterium DJ02. Heilongjiang Anim. Sci. Vet. Med. 2017, 183–186. [Google Scholar] [CrossRef]
- Cao, S.; Chen, M.; Hou, X.; Sun, J. Effect of Novosphingobium sp. Pretreatment on the Structure and Enzymolysis of Two Kinds of Wood. China For. Prod. Ind. 2022, 59, 9–13. [Google Scholar] [CrossRef]
- Zhao, C.; Xie, S.; Pu, Y.; Zhang, R.; Huang, F.; Ragauskas, A.J.; Yuan, J.S. Synergistic Enzymatic and Microbial Lignin Conversion. Green Chem. 2016, 18, 1306–1312. [Google Scholar] [CrossRef]
- Wu, D.; Wei, Z.; Mohamed, T.A.; Zheng, G.; Qu, F.; Wang, F.; Zhao, Y.; Song, C. Lignocellulose Biomass Bioconversion during Composting: Mechanism of Action of Lignocellulase, Pretreatment Methods and Future Perspectives. Chemosphere 2022, 286, 131635. [Google Scholar] [CrossRef]
- Laranjo, M.; Alexandre, A.; Oliveira, S. Legume Growth-Promoting Rhizobia: An Overview on the Mesorhizobium Genus. Microbiol. Res. 2014, 169, 2–17. [Google Scholar] [CrossRef]
- Grieneisen, L.E.; Charpentier, M.J.E.; Alberts, S.C.; Blekhman, R.; Bradburd, G.; Tung, J.; Archie, E.A. Genes, Geology and Germs: Gut Microbiota Across a Primate Hybrid Zone are Explained by Site Soil Properties, not Host Species. Proc. R. Soc. B Biol. Sci. 2019, 286, 20190431. [Google Scholar] [CrossRef]
- Shao, Z.; Chen, X.; He, B.; Wu, L.; Jin, E.; Chen, J.; Wang, S.; Yang, W. Research Progress on Escherichia coli Infections. Anim. Breed. Feed. 2021, 20, 5–7. [Google Scholar] [CrossRef]
- Yan, M. The Mass Diseases of Captive Musk Deer. J. Econ. Anim. 2016, 20, 112–117. [Google Scholar] [CrossRef]
- Nagano, Y.; Itoh, K.; Honda, K. The Induction of Treg Cells by Gut-Indigenous Clostridium. Curr. Opin. Immunol. 2012, 24, 392–397. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg Induction by a Rationally Selected Mixture of Clostridia Strains from the Human Microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Pires, P.S.; Santos, R.L.; da Paixão, T.A.; de Oliveira Bernardes, L.C.; de Macêdo, A.A.; Gonçalves, L.A.; de Oliveira Júnior, C.A.; Silva, R.O.S.; Lobato, F.C.F. Intracellular Survival of Clostridium Chauvoei in Bovine Macrophages. Vet. Microbiol. 2017, 199, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gillis, C.C.; Hughes, E.R.; Spiga, L.; Winter, M.G.; Zhu, W.; Furtado de Carvalho, T.; Chanin, R.B.; Behrendt, C.L.; Hooper, L.V.; Santos, R.L.; et al. Dysbiosis-Associated Change in Host Metabolism Generates Lactate to Support Salmonella Growth. Cell Host Microbe 2018, 23, 54–64.e6. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Zhang, K.; Ma, X.; He, P. Clostridium Species as Probiotics: Potentials and Challenges. J. Anim. Sci. Biotechnol. 2020, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Avram-Hananel, L.; Stock, J.; Parlesak, A.; Bode, C.; Schwartz, B. E Durans Strain M4-5 Isolated from Human Colonic Flora Attenuates Intestinal Inflammation. Dis. Colon Rectum 2010, 53, 1676–1686. [Google Scholar] [CrossRef]
- Hanchi, H.; Mottawea, W.; Sebei, K.; Hammami, R. The Genus Enterococcus: Between Probiotic Potential and Safety Concerns—An Update. Front. Microbiol. 2018, 9, 1791. [Google Scholar] [CrossRef]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.; Ge, J. Comparison of the Gut Microbiota Composition between Wild and Captive Sika Deer (Cervus Nippon. Hortulorum) from Feces by High-Throughput Sequencing. AMB Express 2017, 7, 212. [Google Scholar] [CrossRef]
- Deng, Y.; Peng, J.; Peng, J.; Gao, H.; Huang, P.; Sun, N.; Wang, X.; Yuan, X.; Zhou, H.; Zhang, C. Preliminary Investigation on Feeding Habits of Moschus berezovskii in Jinfo Mountain National Nature Reserve in Chongqing. J. Chongqing Norm. Univ. 2020, 37, 27–34. [Google Scholar]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, M.B.H.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut Microbiota Composition Correlates with Diet and Health in the Elderly. Nature 2012, 488, 178–184. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions across Mammalian Phylogeny and within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.-Q.; Bao, H.; La, N.-T.; Jiang, G.-S.; Zhai, P.-H.; Liu, C.-B.; Yu, L. Gut Microbiota Contribute to Cold Adaptation in Mammals—Primates and Ungulates. iScience 2025, 28, 112245. [Google Scholar] [CrossRef] [PubMed]
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef]
- Jiang, F.; Gao, H.; Qin, W.; Song, P.; Wang, H.; Zhang, J.; Liu, D.; Wang, D.; Zhang, T. Marked Seasonal Variation in Structure and Function of Gut Microbiota in Forest and Alpine Musk Deer. Front. Microbiol. 2021, 12, 699797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wang, X.; Gong, X.; Sui, J. Gut Microbiome Differences in Rescued Common Kestrels (Falco tinnunculus) Before and After Captivity. Front. Microbiol. 2022, 13, 858592. [Google Scholar] [CrossRef]
- Franz, M.; Whyte, L.; Atwood, T.C.; Laidre, K.L.; Roy, D.; Watson, S.E.; Góngora, E.; McKinney, M.A. Distinct Gut Microbiomes in Two Polar Bear Subpopulations Inhabiting Different Sea Ice Ecoregions. Sci. Rep. 2022, 12, 522. [Google Scholar] [CrossRef]
- Tang, S.; Li, Y.; Huang, C.; Yan, S.; Li, Y.; Chen, Z.; Wu, Z. Comparison of Gut Microbiota Diversity Between Captive and Wild Tokay Gecko (Gekko gecko). Front. Microbiol. 2022, 13, 897923. [Google Scholar] [CrossRef]
- Wang, H.; Ali, M.; Zhu, Y.; Chen, X.; Lu, D.; Liu, Y.; Li, K.; Zhang, C. Comparative Analysis of Gut Microbiota in Free Range and House Fed Yaks from Linzhou County. Sci. Rep. 2025, 15, 14317. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Xiao, L.; Liu, Z.; Deng, Y.; Zhu, J.; Yang, C.; Liu, Q.; Tian, D.; Cui, X.; Peng, J. Impacts of Captive Domestication and Geographical Divergence on the Gut Microbiome of Endangered Forest Musk Deer. Animals 2025, 15, 1954. https://doi.org/10.3390/ani15131954
Liu H, Xiao L, Liu Z, Deng Y, Zhu J, Yang C, Liu Q, Tian D, Cui X, Peng J. Impacts of Captive Domestication and Geographical Divergence on the Gut Microbiome of Endangered Forest Musk Deer. Animals. 2025; 15(13):1954. https://doi.org/10.3390/ani15131954
Chicago/Turabian StyleLiu, Huilin, Lu Xiao, Zhiqiang Liu, You Deng, Jinpeng Zhu, Chengzhong Yang, Qing Liu, Di Tian, Xiaojuan Cui, and Jianjun Peng. 2025. "Impacts of Captive Domestication and Geographical Divergence on the Gut Microbiome of Endangered Forest Musk Deer" Animals 15, no. 13: 1954. https://doi.org/10.3390/ani15131954
APA StyleLiu, H., Xiao, L., Liu, Z., Deng, Y., Zhu, J., Yang, C., Liu, Q., Tian, D., Cui, X., & Peng, J. (2025). Impacts of Captive Domestication and Geographical Divergence on the Gut Microbiome of Endangered Forest Musk Deer. Animals, 15(13), 1954. https://doi.org/10.3390/ani15131954