1. Introduction
Mongolian cattle are ancient breeds with a long history of tolerance to cold and roughage, resistance to disease, and adaptation to harsh environments [
1]. Native to the Mongolian Plateau, Mongolian cattle were once prevalent across the Inner Mongolia Autonomous Region and the northeast, northwestern, and southern provinces of China. The most famous breed is found in Ujimqin, Xilingol League, in Inner Mongolia. The Inner Mongolia Autonomous Region is an important production area for Mongolian cattle, which inhabit vast grasslands. Mongolian cattle have traditional husbandry practices. Traditionally, they are mainly raised through year-round grazing, relying on natural grassland resources [
2]. To address over-grazing and promote sustainable grassland management, recent years have seen an increasing number of Mongolian cattle being raised through total mixed ration housed feeding systems. Although the housed feeding method makes full use of resources like corn stalks, silage, and other crop residues and relieves pasture pressure to a certain extent, grazing is still the main feeding method for Mongolian cattle. It enables the cattle to feed on a variety of pasture grasses in their natural environment, making full use of plant fibers and promoting the ecological and forage sustainability of the balance between grass and livestock.
The rumen is a rich and diverse microbial ecosystem that primarily consists of anaerobic bacteria, protozoa, fungi, archaea, and phages [
3]. Rumen microbes are integral to fiber feed breakdown and nutrient transformation, and they also play an extremely important role in the digestive process of ruminants [
4]. These microbiota break down cellulose via fermentation into volatile fatty acids (VFAs), such as acetate, propionate, and butyrate, which are the main sources of energy for ruminants [
5]. Additionally, the ability of the rumen to digest fiber-rich feeds and convert them into various metabolites is an important aspect. These metabolites are utilized by microbiota. Meanwhile, they are also absorbed by ruminants for the maintenance of their growth. Specifically, these processes represent the connection among the host, rumen microbiota, and ration levels. Many researchers have focused on the fact that rumen microbes and metabolites in ruminants are affected by changes in the type of forage and the environment. He et al. [
6] demonstrated that compared with yaks fed full corn silage and supplemented with 3 kg concentrate per head per day, the natural grazing group of yaks feeding on natural forage had higher rumen microbial diversity. Fu et al. [
7] demonstrated that compared with Tan sheep fed a concentrate-supplemented diet, the grazing group of Tan sheep consuming natural forage had higher rumen cellulolytic bacteria. It was also shown [
8] that the rumen fermentation parameters, microbial composition, and metabolome of yaks fed diets with concentrate-to-forage ratios of 50:50, 65:35, and 80:20 have all undergone significant changes. Additionally, Liu et al. [
9] revealed that yak rumen bacterial communities and metabolomes exhibit distinct responses to feed type shifts, with forage-based diets enhancing the abundance of fiber-degrading genera and modulating metabolic pathways related to protein digestion, purine metabolism, and fatty acid biosynthesis.
However, there is still a scarcity of reports on the rumen microbiota and its cellulose degradation function in Mongolian cattle grazing during winter. Most previous studies [
2,
10] on Mongolian cattle have focused on other aspects, such as rumen functional development and microbial succession rather than specifically addressing the rumen microbiota and cellulose degradation function in winter-grazing Mongolian cattle. Therefore, in this study, metagenomics and Liquid Chromatography–Mass Spectrometry (LC-MS) nontargeted metabolomic techniques were used to assess rumen microbial communities, digestive characteristics, and metabolites of Mongolian cattle under grazing natural hay and housed feeding Total Mixed Ration (TMR). It was hypothesized that the two feeding systems, grazing and housed feeding, would regulate the structure of rumen microbial communities and the expression of functional genes, thereby influencing the degradation efficiency of fibrous feed and the composition of metabolites in Mongolian cattle, and ultimately leading to differences in rumen fermentation characteristics and energy utilization directions.
2. Materials and Methods
2.1. Animal and Experiment Design
In total, 12 healthy local Mongolian female cattle (3–4 years old; weighed 460 ± 35 kg) were selected as test animals in Uragai Ranch, Xilingol League, Inner Mongolia Autonomous Region. These cattle were divided into a housed feeding group (S group) and a grazing group (F group). Each group contained six animals, and a completely randomized experimental design was adopted. The F group of Mongolian cattle grazed in the Uragui grasslands and fed on natural pasture (the main types of pasture are
Leymus chinensis,
Filipendula ulmaria,
Festuca capillata, etc.). Mongolian cattle in the S group were kept in semi-open pens and fed a basal ration. The basal ration was formulated according to NRC guidelines [
11] (
Table S1). The experiment was conducted in winter, with a 10-day pretrial period followed by a 90-day formal experimental period. The Mongolia cattle in the S group were fed with 12 kg of TMR every day. All animals were free to feed and drink.
At the conclusion of this experiment, rumen fluid was collected via a flexible oral gastric tube equipped with a metal filter prior to the morning feeding. A vacuum rumen tube (Wuhan Colibri Ranching Technology Co., Ltd., Wuhan, China) was used to collect rumen fluid through the mouth, and 200 mL of rumen fluid was first extracted and discarded. Then, 200 mL of rumen fluid was extracted, filtered through four layers of sterilized gauze, and aliquoted into 15 mL cryopreservation tubes, which were stored in a liquid nitrogen tank and then sent to Meiji Biologicals, Inc. (Tokyo, Japan), within 2–4 h with dry ice for microbial DNA extraction and sequencing analysis.
2.2. Determination of Feed Nutrient Content
The levels of crude protein (CP; method 2001.11), neutral detergent fiber (NDF; method 2002.04), acid detergent fiber (ADF; method 973.18) and starch (starch; method 996.11) were measured following the procedures of AOAC (2005) [
12]. Calcium and phosphorus (P) in feed samples were analyzed using inductively coupled plasma mass spectroscopy (AOAC, 2005; method 985.01) [
12].
2.3. VFA and Cellulose Degrading Enzyme Activity Index Measurements
pH: The pH of the rumen fluid was determined using a PHSJ-3F laboratory pH meter (Yantai Stark Instrument Co., Ltd., Yantai, China). Before measurement, the pH meter was calibrated with standard buffer solution. After taking the rumen fluid out of storage and allowing it to thaw completely, the electrode was immersed in the thawed rumen fluid, and the pH was recorded after the reading stabilized.
VFAs: The concentration of VFAs was determined using an Agilent 1160 gas chromatograph following the method described by Erwin et al. [
13] (Agilent Technologies, Santa Clara, CA, USA).
Cellulose-degrading enzyme activity: Following the method described by Guo et al. [
14], a kit from Jiangsu Enzyme Immunity Industry Co., Ltd. (Nanjing, China) was used. Ruminal digestive enzymes (cellulase, xylanase, and β-glucosidase) were measured via ELISA.
2.4. DNA Extraction and Macrogenomic Determination
The rumen fluid obtained above was used for microbial DNA extraction using an MP kit (MP Biomedicals, Santa Ana, CA, USA). DNA concentration and purity were measured using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The extracted DNA then underwent quality testing, fragmentation, and end repair to construct a sequencing library. The constructed library was subjected to quality and concentration control using agarose gel electrophoresis and qPCR, mixed with DNA polymerase and fluorescently labeled primers, and sequenced for DNA fragments on the Nova Seq platform; the sequencing was performed by Meiji Bio (NovaSeq platform, Shanghai, China).
2.5. Macrogenome Sequencing and Analysis of Rumen Microorganisms
The fragments from sequencing were filtered using the Fastp software (v 0.20.0) to obtain high-quality clean reads for subsequent analysis. The sequences were spliced and assembled using the software MEGAHIT (v 1.1.2) [
15], followed by open read frame (ORF) prediction of the contigs using Prodigal (v 2.6.3). Genes with nucleic acid lengths of 100 bp or more were selected and converted into amino acid sequences. Nonredundant gene collections were created using the CD-HIT (v 4.6.1, default parameters: 90% identity, 90% coverage) [
16] software to obtain the base sequences of the genes in the nonredundant gene sets. Next, SOAPaligner (v 2.21) was used to compare high-quality reads with nonredundant gene sets for each sample to collect data on gene prevalence across samples. All online databases and software were accessed in August 2023. Information on species composition was obtained using the software BLASTP (v 2.2.28+,
http://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 2 August 2023) compared to the NR database (
https://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/, accessed on 4 August 2023). The Kyoto Encyclopedia of Genes and Genomes (KEGG) database (
https://www.genome.jp/kegg, accessed on 8 August 2023) was compared using the software BLASTP (v 2.2.28+,
http://blast.ncbi.nlm.nih.gov/Blast.cgi, accessed on 10 August 2023) to obtain the corresponding KEGG annotation profiles of the genes and perform statistical analysis. An annotation profile of carbohydrate-active enzyme-encoding genes was obtained using hmmscan against the CAZy database (
http://www.cazy.org/, accessed on 14 August 2023).
2.6. Non-Targeted Metabolome Sequencing
A sample of rumen fluid was collected in a centrifuge tube, injected with a water-acetonitrile mixture, vortexed and mixed for 30 s, and left undisturbed in an ice bath before centrifugation was performed. Simultaneously, an equal volume of the sample was mixed to make a quality control (QC) sample. The samples were separated using a Hypesil Gold column (manufactured by Thermo Fisher Scientific, Waltham, MA, USA) in positive and negative ion modes and screened for particles with mass-charge ratios between 70 and 1000 using a Q ExactiveTM HF-X mass spectrometer (manufactured by Thermo Fisher Scientific, Waltham, USA) in positive and negative ion scanning modes. After finalizing the onboarding process, the unprocessed data were analyzed using the Progenesis QI software (v 2.0, Waters Corporation, Milford, MA, USA). In this step, variables from the quality control samples that had a relative standard deviation (RSD) greater than 30% were filtered out, resulting in refined data matrix postprocessing. The mass spectral information was matched with HMDB and Metlin as well as Meggie to construct libraries. Information on metabolites was obtained. The processed matrices were submitted to the Megibio cloud platform (
https://cloud.majorbio.com, accessed on 4 August 2023) for evaluation, and the data were processed and analyzed using the partial least squares discriminant analysis (PLS-DA) software, metaX (v 1.4.2), to obtain the variable importance projection (VIP), retention time (RT), and mass-to-charge ration (m/z) of the metabolites. Statistical analysis of metabolites between the two groups was conducted to calculate the multiplicity of differences in metabolites between different groups [
17]. VIP > 1.0,
p < 0.05, FC > 1.5, or FC < 0.667 were used as the screening criteria to identify the differential metabolites, and the pathway information on metabolite involvement was obtained according to the KEGG database (
https://www.kegg.jp/, accessed on 16 August 2023) [
18]. Pathway enrichment was conducted using the Python software package (v 3.0) and the key biological pathways related to the experimental treatments were identified by conducting Fisher’s exact test.
2.7. Statistical Analysis
Prior to statistical analysis, the normality of rumen fermentation parameters, cellulase activity, and α diversity index was assessed using the Shapiro–Wilk test. Homogeneity of variances was evaluated using Levene’s test. All parameters satisfied the assumptions of normality (Shapiro–Wilk p > 0.05) and homogeneity of variances (Levene’s p > 0.05). Statistical differences were analyzed using an independent sample t-test via SPSS 27.0 (IBM, Armonk, NY, USA). Significance was set at p < 0.05, and results were presented as the mean ± standard error of the mean (SEM).
The rumen microbial phyla and genera were compared using the Welch t-test, with the FDR adjusted p < 0.05 being considered as significantly different. The KEGG pathways and CAZymes were compared by the Wilcoxon rank-sum test, with the FDR adjusted p < 0.05 being considered significantly different. Using PLS-DA and Student’s t-test, the significance of metabolite difference in each group was analyzed, and the difference was statistically significant with p < 0.05. Pathway enrichment was conducted using the Python software package and the difference was statistically significant with p < 0.05. The correlations between different metabolites and bacterial communities were calculated by Pearson correlation analysis using the R program (v 3.6.1), and p < 0.05 indicated significant differences.
4. Discussion
Ruminal pH is an important indicator used to evaluate the health of the rumen and visualize the internal setting of the rumen, which is affected by the combination of feed type, ration intake, saliva secretion, and the accumulation of organic acids [
19]. VFA produced by microbiota in the rumen through fermentation is a key form of energy for ruminants, as they provide 70% of metabolic energy [
5]. Microbiota initially degrade crude fiber, starch, and soluble sugars into pyruvic acid, which is then converted into various VFAs (acetate, propionate, butyrate, isobutyrate, valerate, and isovalerate) through distinct metabolic pathways [
20]. Previous research [
21,
22] have indicated that an increased presence of cellulose, hemicellulose, and lignin in feedstuffs enhances the production of acetate during rumen fermentation. Furthermore, as acetate constitutes a major portion of TVFA, its rise contributes to an overall increase in TVFA levels. In the present study, the KEGG pathway analysis revealed that carbohydrate and pyruvate metabolism were significantly upregulated in the F group, indicating that a greater amount of crude fiber serves as a substrate for microbial fermentation and VFA synthesis in Mongolian cattle under grazing conditions. Consequently, in comparison to the S group, the F group exhibited higher TVFA levels and a marked increase in acetate concentration. Although the two feeding systems, i.e., grazing and housing, affected the rumen pH, currently, we could only observe that the rumen pH was maintained within a certain range, but a more precise definition of this range and the measurement of the buffer capacity of the rumen microbiota will be the focus of our follow-up research to better understand its adaptability to the different feeding modes and the stability of the internal environment of the rumen.
The rumen ecosystem is focused primarily on the breakdown of plant fibers through a diverse community of microbiota. This function allows ruminants to digest feed, thus emphasizing its importance for human needs, as it is the main contributor to the conversion of plant biomass into essential products (milk, meat, and fiber products) [
23]. Plant cell walls are composed of biopolymers and structural proteins such as cellulose, hemicellulose, lignin, and pectin, with cellulose being the main component in plant cell walls [
24]. Cellulases produced by rumen fibrolytic bacteria can cleave the β-1,4 bonds in the cellulose chain, thus degrading cellulose polymers [
25]. β-Glucosidase can effectively degrade cellobiose and cellotriose [
26] and convert cellulose into available glucose. Xylanases are an important class of hemicellulases that play a key role in breaking down the complex structure of xylan [
27]. These abundant CAZymes facilitate the efficient degradation of cellulose. It has been reported [
28] that the supplementation of isovalerate enhances cellulase activity and leads to an increase in total rumen VFA concentration in calves. In this study, isovalerate, cellulase, xylanase, and β-glucosidase (β-GC) levels were significantly higher in the F group compared to those in the S group, indicating that Mongolian cattle can stimulate cellulase activity, accelerate the fixation of cellulase, and enhance the ability to digest cellulose and hemicellulose under grazing conditions.
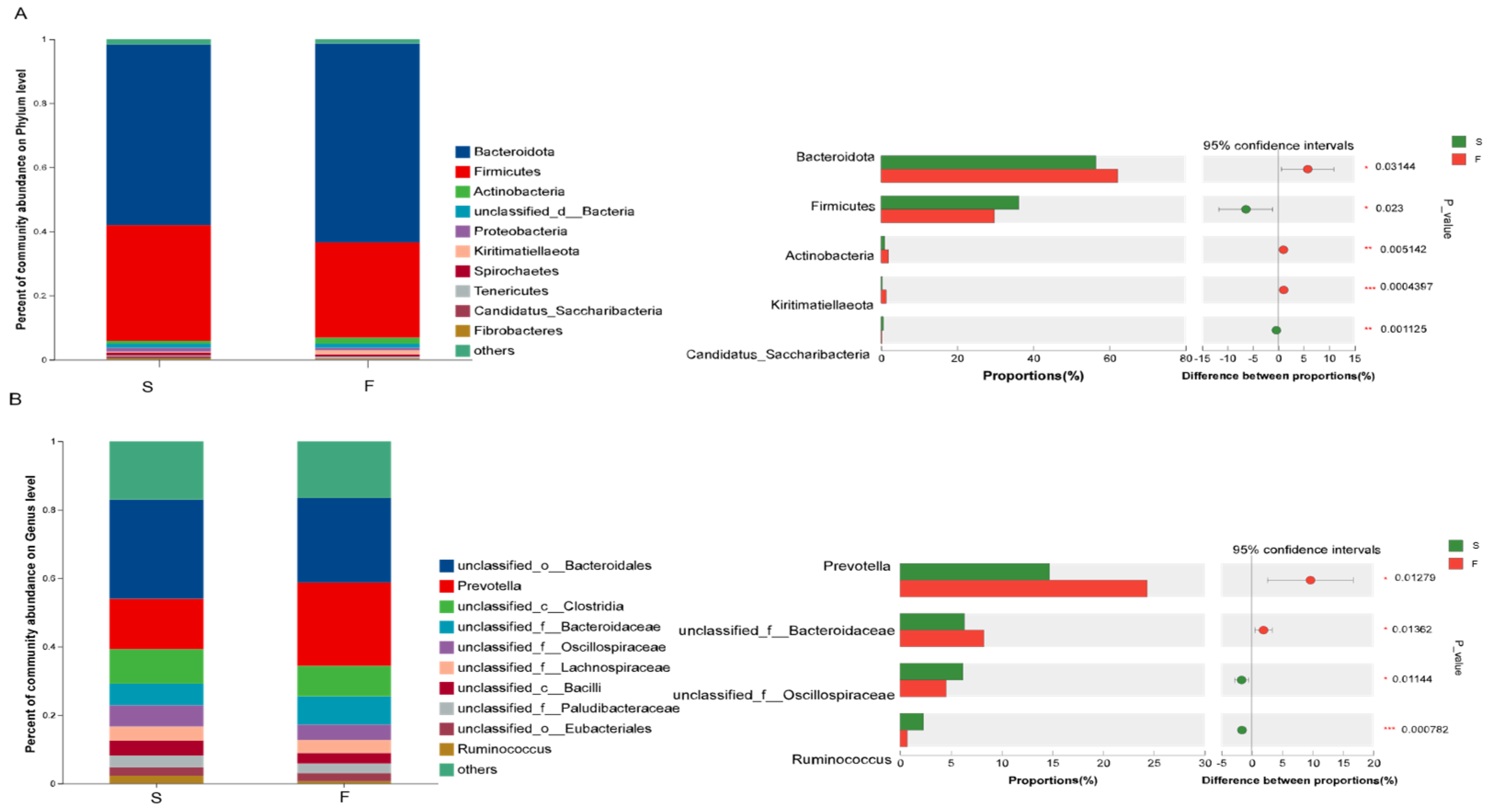
The sequencing results revealed that different feeding systems affected the composition of the rumen bacterial community. Ruminal microbiota play an extremely important role in ruminants by breaking down cellulose and hemicellulose into simple sugars and short-chain fatty acids through fermentation. Rumen bacteria play a key role in the degradation of cellulose and hemicellulose [
25]. Many studies in ruminants [
9,
29], have shown that the dominant phyla of rumen microbiota are Bacteroidota and Firmicutes, regardless of their ration composition. The results of this experiment revealed that Bacteroidota and Firmicutes were the dominant phyla in the rumens of Mongolian cattle in all the samples from the different feeding systems. Some studies [
30] have found that Bacteroidota can degrade crude cellulose, but Bacteroidota may be affected by carbohydrate structure. Firmicutes are the main degraders of complex carbohydrates [
31]. Studies [
9,
32], have also found that Firmicutes significantly increased and Bacteroidota significantly decreased in the rumen microbiota of ruminants as the level of concentrate in the feed increased. The results of this study revealed that the abundance of Bacteroidota significantly increased in the rumen of the Mongolian cattle in the F group (higher crude fiber content in the Mongolian cattle in the F group) and that the abundance of Firmicutes significantly decreased compared to that in the S group, which matched the results of the above study. The reasons for these changes are that in the grazing system, pastures contain large amounts of cellulose, hemicellulose, and structural carbohydrates which are difficult to ferment. Therefore, Mongolian cattle raised through grazing consume more plant fiber than those raised under housed feeding. These differences have led to changes in the rumen microbial composition, thus confirming the differences in the rumen flora under grazing and housed feeding conditions.
At the genus level,
unclassified__o___Bacteroidales, Prevotella,
unclassified__c__Clostridia, and
unclassified__f__Bacteroidacea were the dominant genera in both groups in this study. Zhou et al. [
32] reported that the relative abundance of
Prevotella was significantly greater in the grazing and supplemental-feeding groups than in the forage-fed group. The relative abundance of
Ruminococcus was lower in the F group than in the forage-fed and supplemental feeding groups.
Prevotella is one of the most representative bacterial genera in the rumen biosystem, and it plays an important role in phytocellulose degradation [
17]. Chiquette et al. [
33] reported that supplementing dairy cows with
Prevotella as a direct-fed microorganism significantly increased acetate concentration in the rumen. This suggests that the higher abundance of
Prevotella in the rumen of Mongolian cattle in the F group may enhance cellulose degradation, leading to the production of greater amounts of acetate. This finding may indirectly explain the elevated acetate concentration observed in the F group. A study [
34] found that
Ruminococcus, a high-level energy group, plays a regulatory role in maintaining the stability of the rumen environment in yaks as the energy content of the ration increases. The results of this study revealed that in the F group, the abundance of
Prevotella in Bacteroidota was increased and the abundance of
Ruminococcus in Firmicutes was decreased. These findings suggested that Mongolian cattle are more capable of degrading cellulose from pastures under grazing conditions and that housed conditions help maintain the stability of the rumen environment, which could potentially have a positive impact on the health of Mongolian cattle.
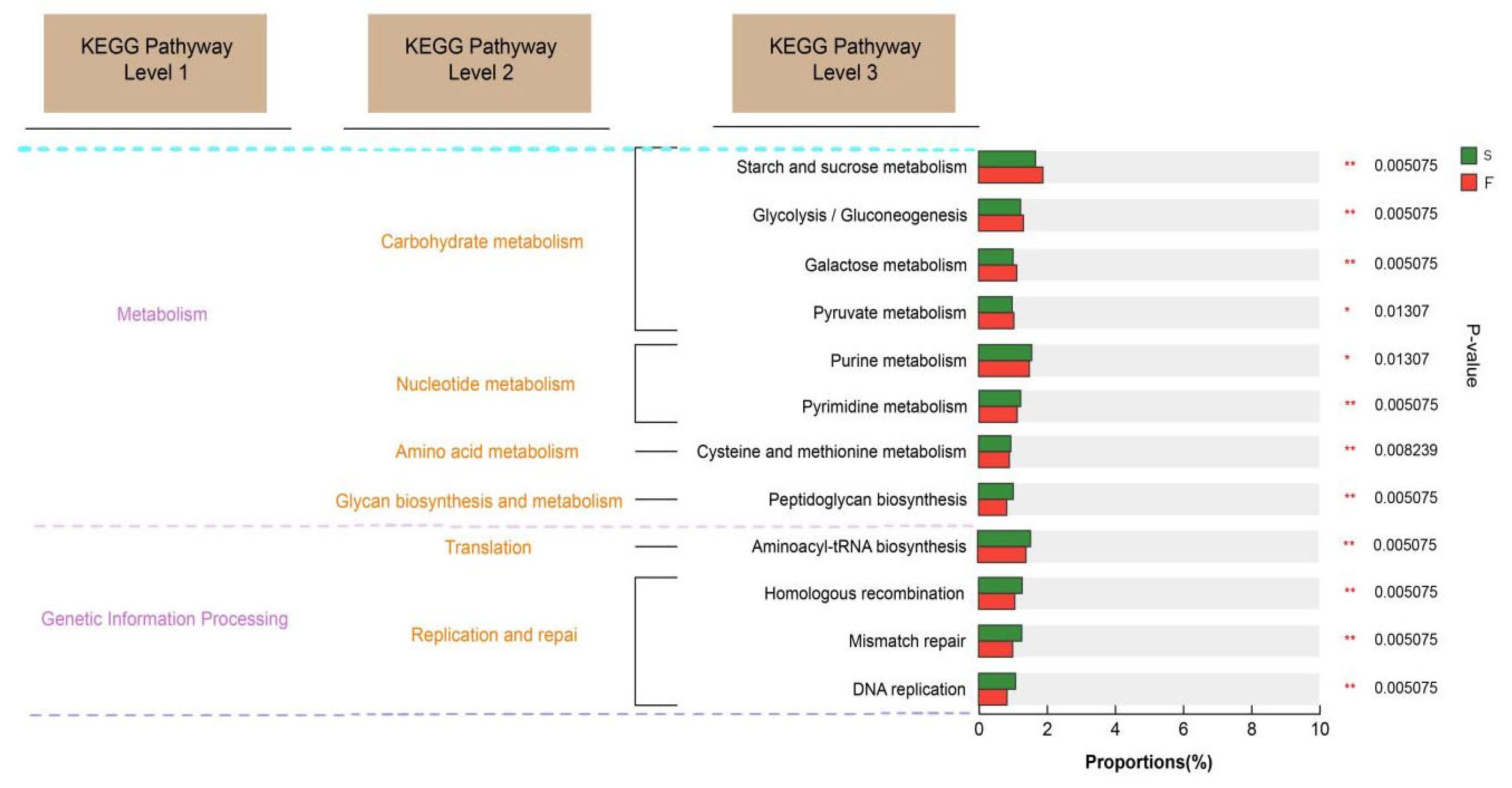
The KEGG database is a comprehensive bioinformatics database that integrates genomic, chemical, and systemic functional information to reveal the genetic material and chemical blueprint of life forms. By annotating the KEGG functions related to rumen microbial function in Mongolian cattle with different feeding systems, we found that the KEGG function related to carbohydrate degradation was enriched in the rumens of the Mongolian cattle in the F group. Glycolytic pathways include starch and sucrose metabolism, galactose metabolism, and the interconversion of glucose and pyruvate. These findings suggested that Mongolian cattle in the F group are more capable of degrading carbohydrates and may produce more hydrolysis products and pyruvate. Mongolian cattle in the S group were enriched mainly in purine metabolism, pyrimidine metabolism, and peptidoglycan biosynthesis, as well as cell growth and death. Purines are abundant in organisms and are key raw materials for cell proliferation and important factors in immunomodulation [
35]. Liu et al. [
9] reported that feeding concentrates are more helpful for purine metabolism in the rumen of yaks. These findings suggest that purine metabolism is increased in the rumen of Mongolian cattle under housed feeding conditions, which may affect their growth and development.
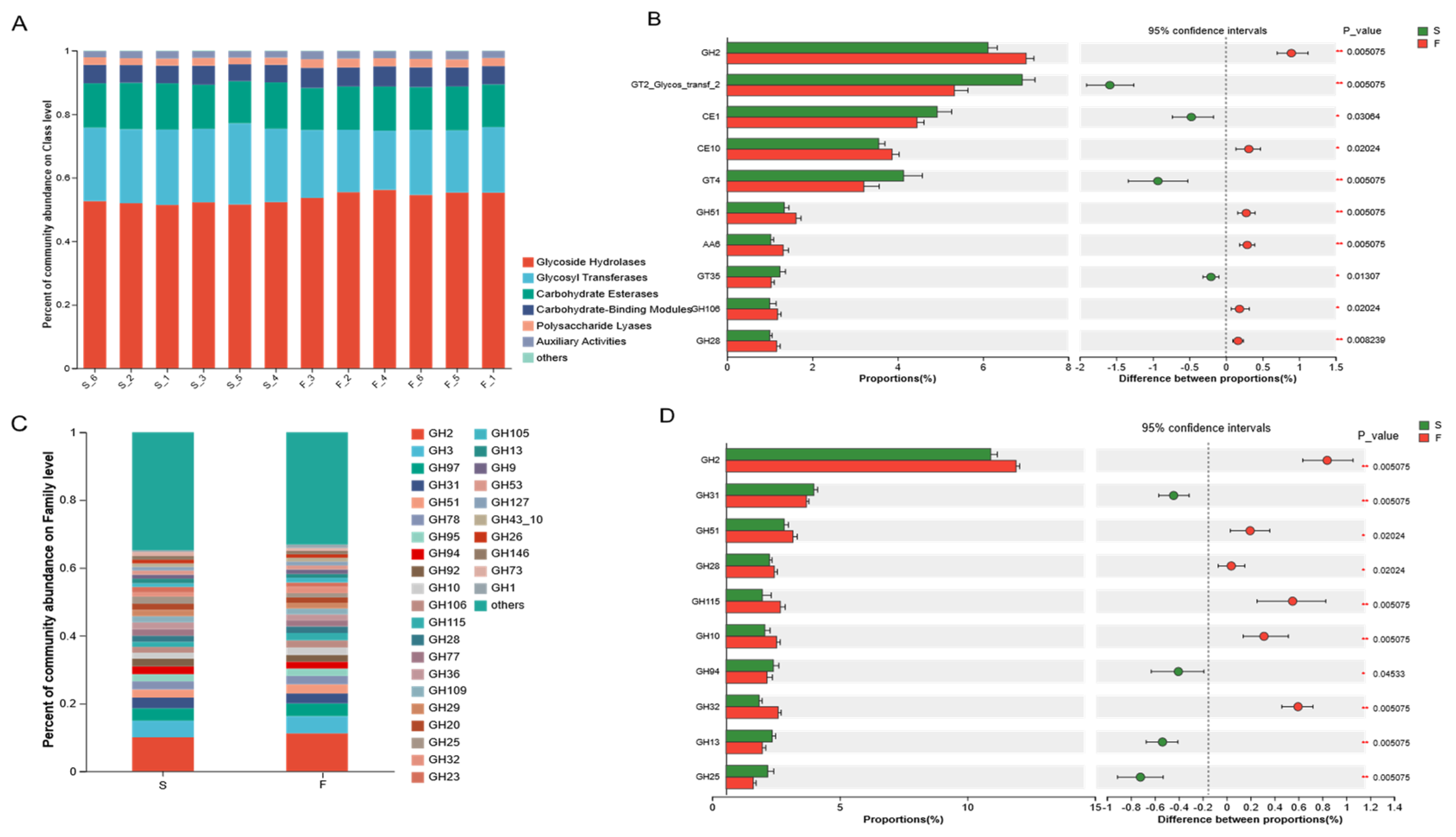
CAZymes is a database resource on enzymes that can synthesize or degrade complex carbohydrates and sugar compounds. Ruminants can break down the cellulose structure of plant cell walls using carbohydrate-active enzymes secreted by microbiota in the rumen [
36]. The synergistic action of multiple GHs on different parts of complex biopolymers facilitates the efficient degradation of plant fibers [
23]. The results of this experiment revealed that the greatest relative abundance of GHs was associated with the F and S groups, accounting for 53.64% of all CAZymes, followed by GTs, CEs, CBMs, PLs, and AAs, reflecting the dominant ability and importance of the GH family in the degradation of fiber. Dai et al. [
37] reported that cellulases belong to GH5, GH9, GH45, and GH48; two-thirds of hemicellulases belong to GH10, GH11, and GH26; and most of the oligosaccharide-degrading enzymes belong to GH1, GH2, GH3, and GH43, as determined by sub-transcriptomic analysis. In this study, Mongolian cattle in the F group presented greater cellulase (GH9), hemicellulase (GH10, GH28), and oligosaccharide-degrading enzyme (GH2) contents and greater xylan hydrolyzing activity enzyme (GH51) than Mongolian cattle in the S group. These findings suggested that, under grazing conditions, Mongolian cattle have a greater ability to degrade pasture, probably not only through enhanced cellulose degradation but also through enhanced hemicellulose degradation. The degradation of hemicellulose facilitates cellulose digestion in forage by exposing cellulose to microbial activity [
38]. Previous studies [
39] have indicated that a high abundance of
Prevotella is typically associated with diets rich in plant-derived glycans and dietary fiber, and that
Prevotella CAZy cluster enzymes, including GH51 and GH28, participate in the galactose degradation pathway. These findings suggest that the microbial community in Mongolian cattle from the F group exhibited an enhanced fiber degradation capacity due to increased expression and secretion of CAZymes involved in plant fiber breakdown. Specifically, the high abundance of the GHs family and the metabolic specialization of
Prevotella allowed Mongolian cattle in the F group to efficiently utilize complex carbohydrates from pasture. This may partially explain the significant increase in galactose metabolism observed in the F group. The diets of Mongolian cattle under-housed conditions are rich in starch; therefore, the starch-degrading enzymes (GH13, GH31) are enriched in the S group. The rumen microbiota can better utilize starch and acquire more energy and other nutrients, which is beneficial to the growth and body maintenance of Mongolian cattle.
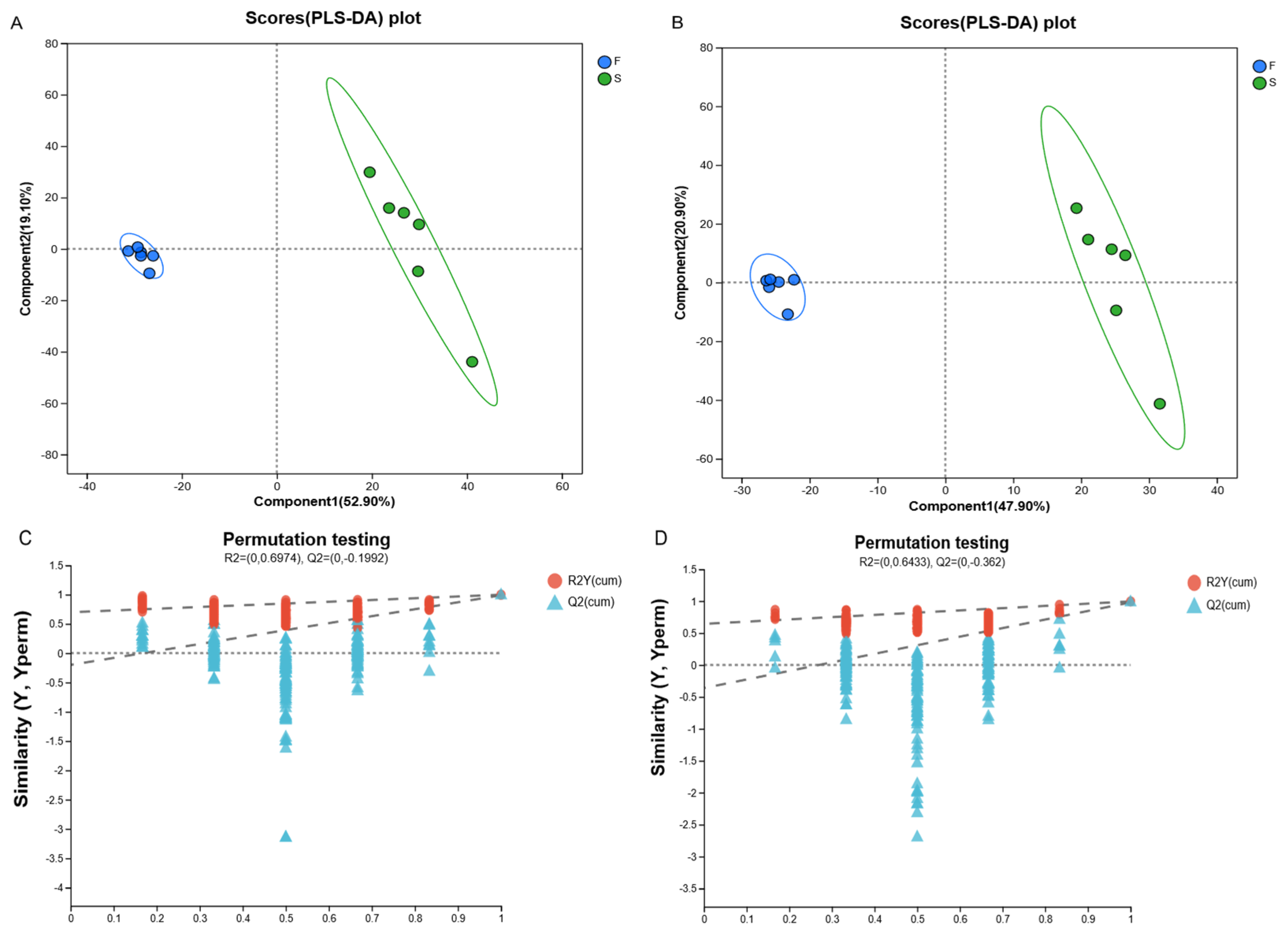
Metabolomics can reveal the role of microbial communities in ruminant digestion, absorption, and energy utilization and better understand host–microbe symbiosis. We evaluated the metabolites in the rumen fluid of Mongolian cattle in the F group and the S group via LC-MS metabolomic analysis and found that the samples from the F group and the S group were significantly separated, as determined by PLS-DA analysis and replacement tests, which confirmed that the different feeding systems significantly affected their rumen metabolism. We investigated the mechanisms underlying the effects of metabolites on host and rumen microbiota in Mongolian cattle under different feeding systems by integrating the rumen fluid metabolome and performing a correlation analysis of the dominant genera of rumen microbiota with differential metabolites. Subaphylline is an intermediate of arginine metabolism in arginine and proline metabolism (KEGG, map00330) and is formed by the combination of putrescine and ferulic acid through an enzyme-catalyzed reaction. Ferulic acid (FA) is a common phenolic acid in nongrain feeds such as pasture and straw, and Ferulic acid esterase (FAE) is an enzyme that catalyzes the ester-bond hydrolysis reaction between FA and acyl substrates, which efficiently releases FA from roughage substrates and promotes their crude fiber degradation ability in concert with other crude fiber-degrading enzymes (e.g., xylanase and cellulase) [
40]. The upregulation of subaphylline in Mongolian cattle in the F group and the role of FAEs in crude fiber degradation in this study suggested that grazing increases the metabolites of digested cellulose in Mongolian cattle. Amino acids not only play important roles in the growth and metabolism of microbiota but are also the basic substances required for many important physiological processes, such as optimal growth, reproduction, normal lactation, and effective maintenance of body proteins in animals. Among organic acids and their derivatives, L-arogenate, an important intermediate metabolite in the biosynthesis of phenylalanine, tyrosine, and tryptophan, was downregulated in the rumens of Mongolian cattle in the F group (KEGG, map00400). Yi et al. [
8] reported that a yak group with a higher ration concentrate ratio had relatively strong rumen amino acid metabolism. These findings suggested that compared to the F group, the S group presented greater rumen amino acid metabolism. To summarize, the metabolic changes were consistent with the metagenome results. The relationship between rumen microbial taxa and host metabolism was revealed by the joint examination of prevailing microbial genera and varying metabolites.
Prevotella, Bacteroidota, and
Ruminococcus were highly significantly associated with metabolites, among which included diglycerides related to lipid metabolism [DG (18:4(6Z,9Z,12Z,15Z)/15:0/0:0), and DG (13:0/20:3 (5Z,8Z,14Z)-O (11S, 12R)/0:0)] were significantly lower in the F group. A study [
41] showed that diglycerides are important intermediates in fat synthesis.
Prevotella, Bacteroidota, DG (18:4 (6Z,9Z,12Z,15Z)/15:0/0:0), and DG (13:0/20:3 (5Z,8Z,14Z)-O (11S,12R)/0:0) all showed highly significant negative correlations, whereas
Ruminococcus was the opposite, suggesting that living conditions can promote fat synthesis in Mongolian cattle. The correlations shown by
Prevotella, Bacteroidota, and
Ruminococcus with these metabolites under different feeding regimes suggested that metabolite levels are influenced by microbiota abundance, thus affecting the regulatory functions of the organism. Among them,
Prevotella and Bacteroidota were the key contributors to the rumen microbial metabolites of the Mongolian cattle in the F group, and
Ruminococcus was the key contributor to the rumen microbial metabolites of the Mongolian cattle in the S group.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}