The Transcriptomic Signature of Donkey Ovarian Tissue Revealed by Cross-Species Comparative Analysis at Single-Cell Resolution

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Animals
2.3. Single Cell Suspension Preparation and Sequencing
2.4. The Quality Control and Preprocessing of scRNA-Seq Data
2.5. Transcriptional Similarity Analysis Between Cell Types Across Species
2.6. Multispecies Correlation Analysis
2.7. Gene Regulatory Network Inference
2.8. Analysis of Differentially Expressed Genes in Cell Clusters
2.9. Protein–Protein Interaction Analysis
2.10. Evaluation of Metabolic Activity
2.11. Single-Cell Developmental Trajectory Construction
2.12. Transcriptional Regulatory Network Analysis
2.13. Cell–Cell Interaction Analysis
2.14. GO and KEGG Enrichment Analysis
2.15. Immunofluorescence Staining
2.16. Cumulus–Oocyte Complex (COC) Collection and Small Interfering RNA (siRNA) Transfection
2.17. Detection of Spindle Assembly
2.18. Reactive Oxygen Species (ROS) and Mitochondrial Membrane Potential Evaluation
2.19. RNA Extraction and Real-Time Quantitative PCR (RT-qPCR)
2.20. Phylogenetic Trees
2.21. Statistical Analysis
3. Results
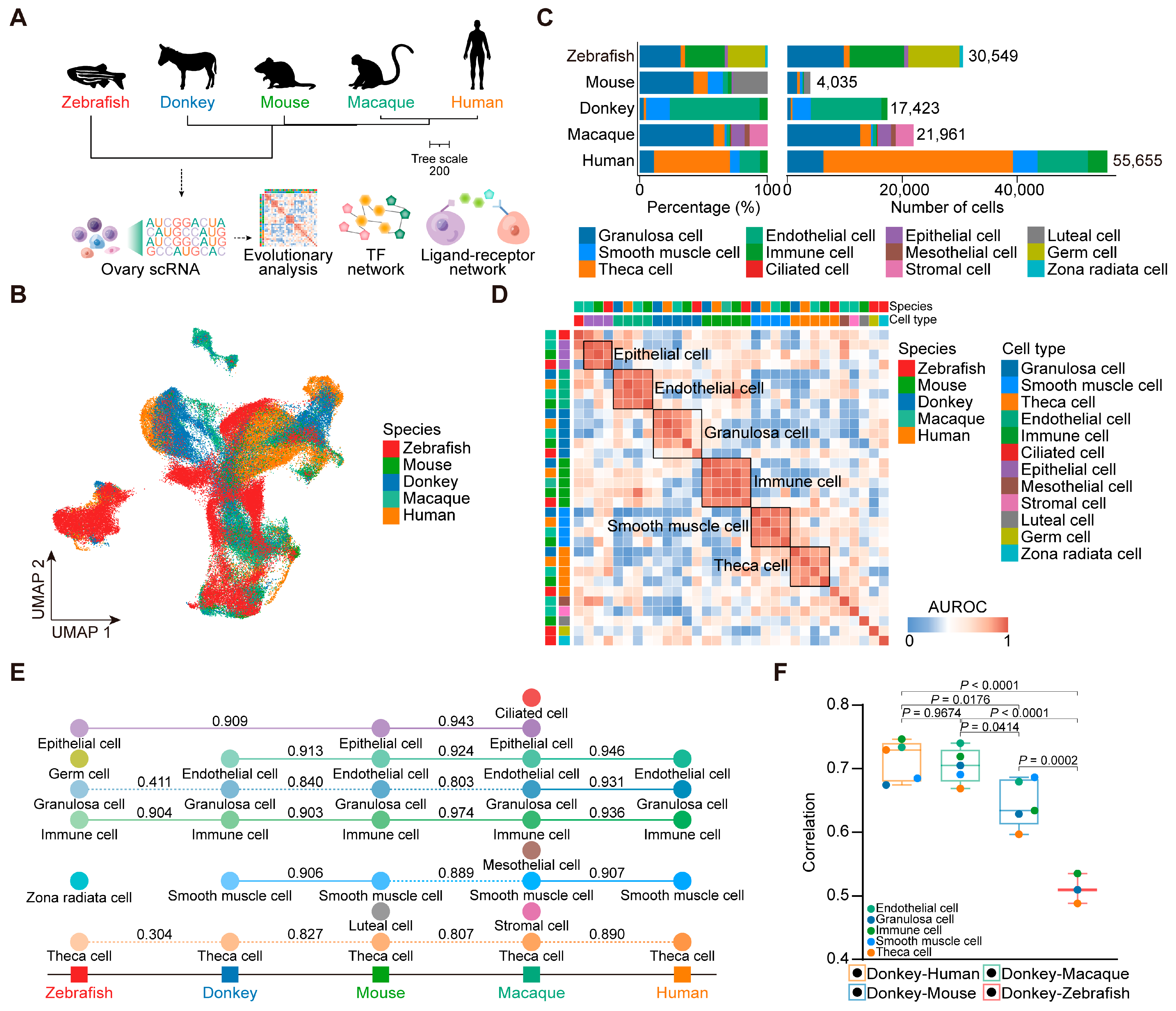
3.1. Identification of Ovarian Cell Types in Dezhou Donkey
3.2. Cross-Species Ovarian Cell Transcriptional Similarity Analysis
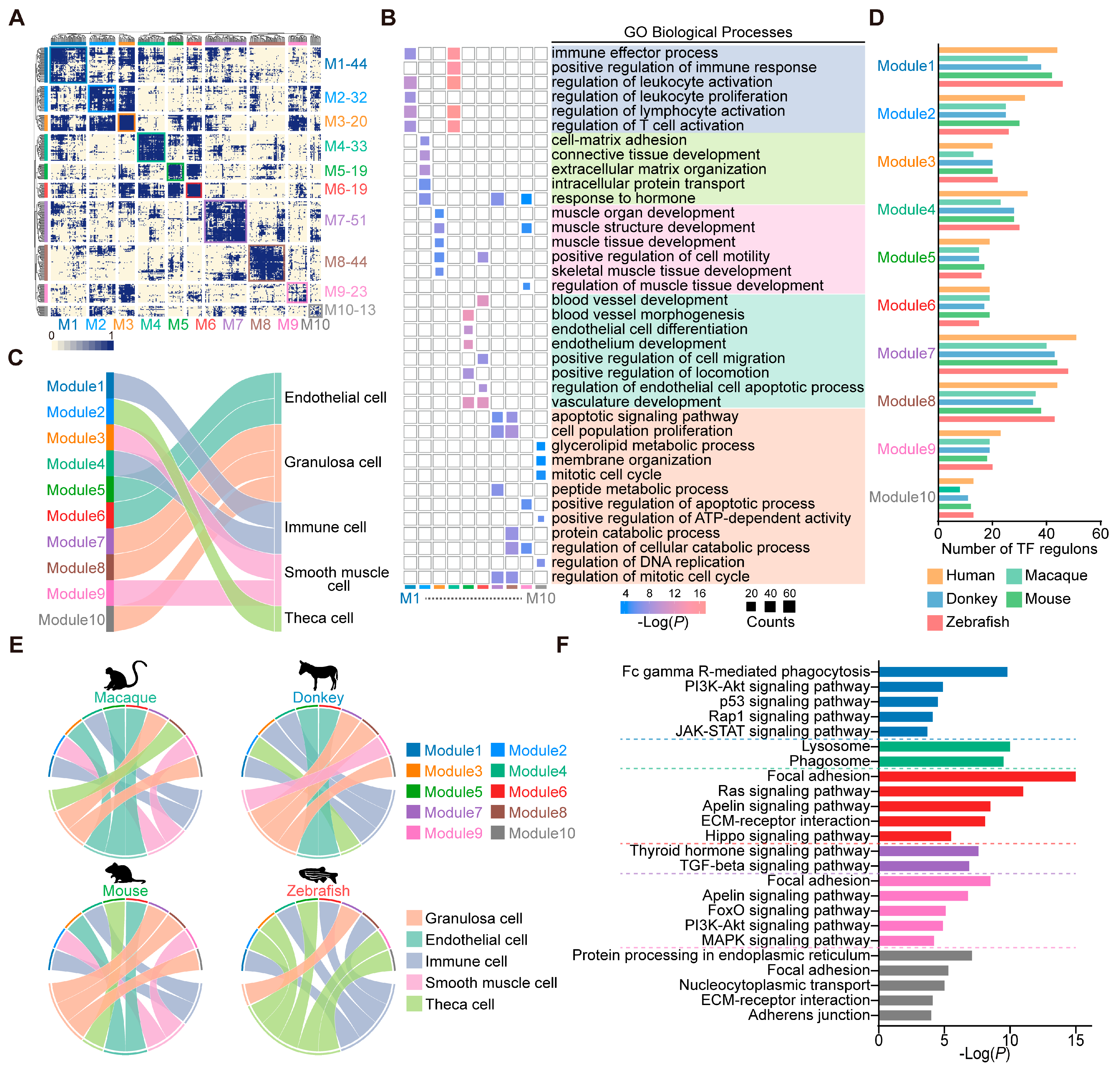
3.3. Conserved Transcription Factor Modules in Ovarian Cells Across Species
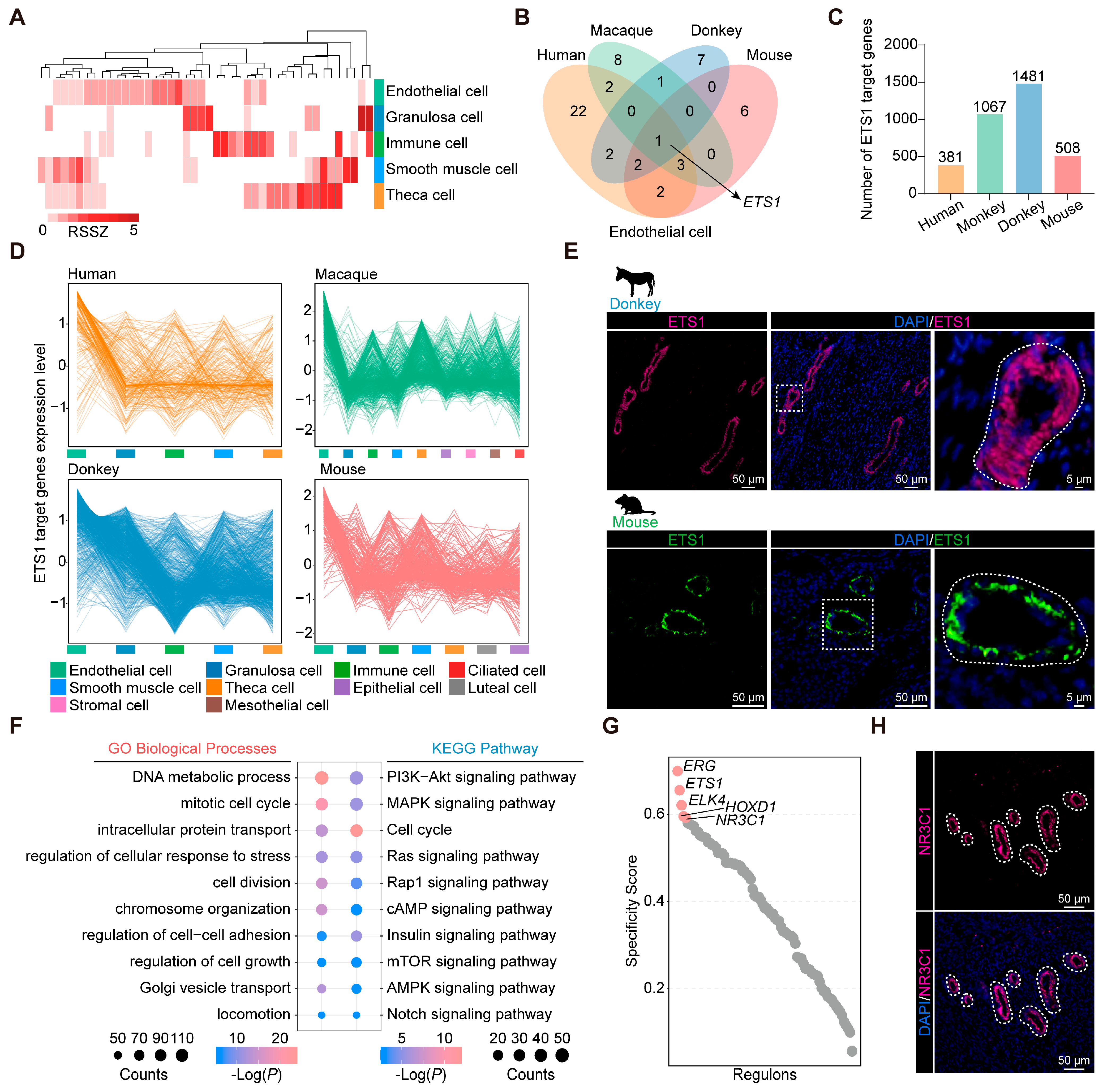
3.4. Identification of Conserved and Characteristic Transcription Factors in Ovarian Endothelial Cells
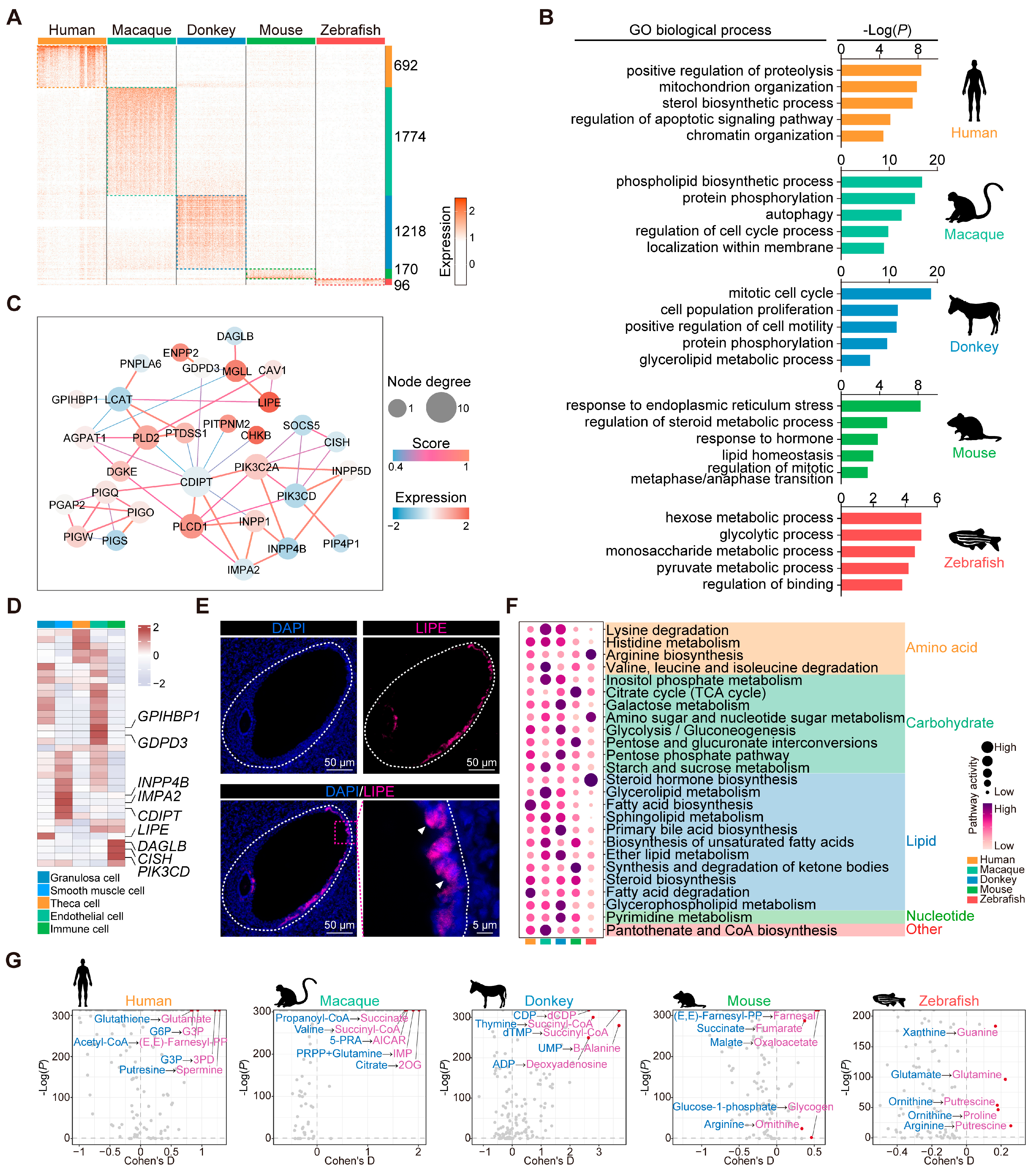
3.5. Conserved Marker and Functional Genes in Granulosa Cells
3.6. Comparative Analysis of Granulosa Cells from Different Species
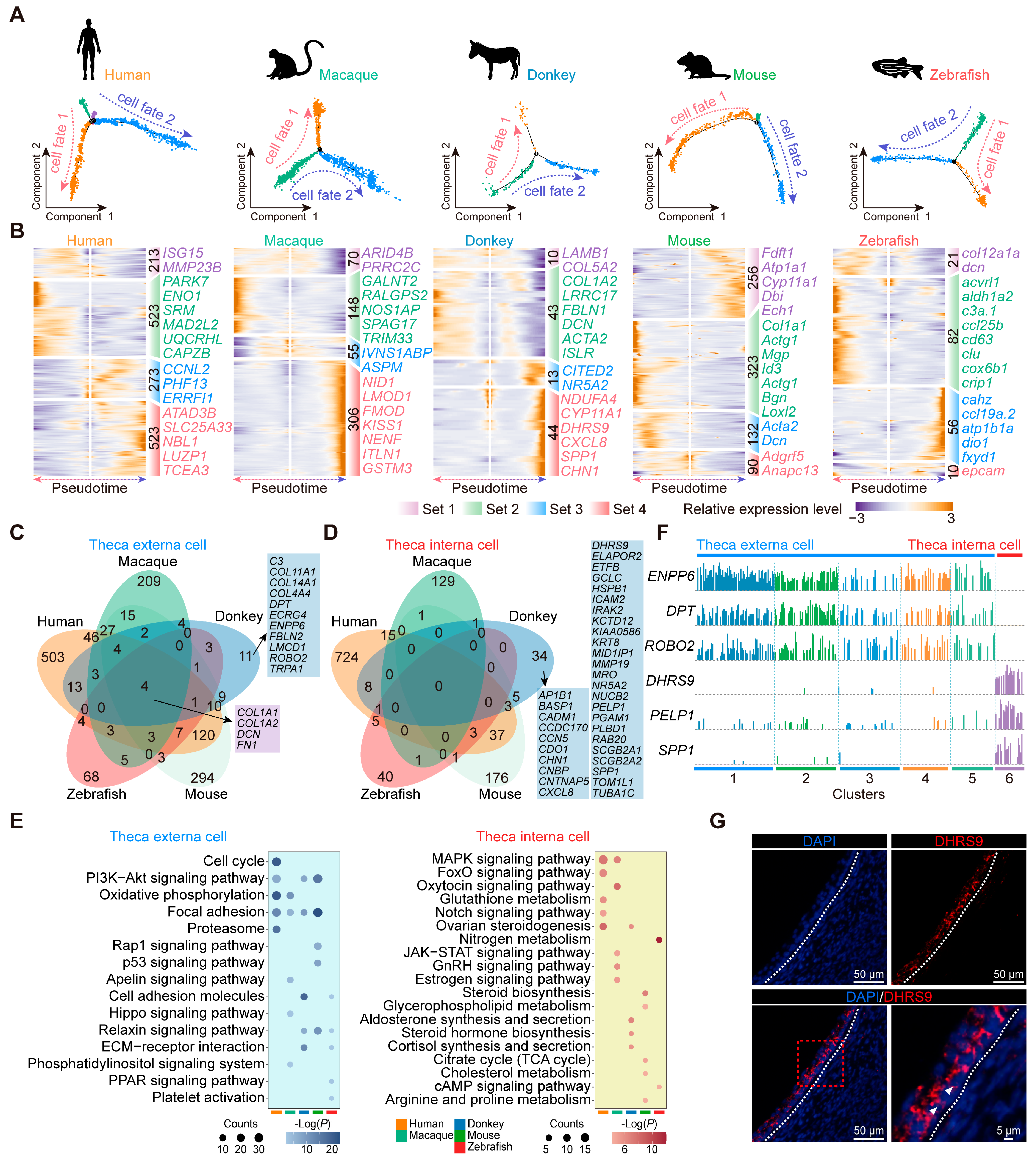
3.7. Conservation and Characteristics of Theca Cell Development in Diverse Species
3.8. Interaction Networks of Ovarian Cells in Each Species
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUROC | Area under the receiver operator characteristic curve |
| BSA | Bovine serum albumin |
| COC | Cumulus–oocyte complex |
| CSI | Connection similarity index |
| ECM | Extracellular matrix |
| GO | Gene Ontology |
| GR | Glucocorticoid receptor |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LPA | Lysophosphatidic acid |
| NC | Negative control |
| PCC | Pearson’s correlation coefficient |
| PFA | Paraformaldehyde |
| PMSG | Pregnant mare serum gonadotropin |
| ROS | Reactive oxygen species |
| RPCA | Reciprocal principal component analysis |
| RSSZ | Z-score normalized regulon specificity score |
| RT-qPCR | Real time quantitative PCR |
| scRNA-seq | Single-cell RNA sequencing |
| SDS–PAGE | SDS–polyacrylamide gel electrophoresis |
| SHMT | Serine hydroxymethyltransferase |
| siRNA | Small interfering RNA |
| T3 | Triiodothyronine |
References
- Wang, Y.; Hua, X.; Shi, X.; Wang, C. Origin, Evolution, and Research Development of Donkeys. Genes 2022, 13, 1945. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Albenzio, M.; Della Malva, A.; Muscio, A.; Sevi, A. Nutritional properties and consumer evaluation of donkey bresaola and salami: Comparison with conventional products. Meat Sci. 2015, 101, 19–24. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhang, Y.; Meng, J.; Wu, M.H.; Ma, Z.G.; Xia, M.; Fu, G.L.; Cao, H. Herbalogical study of Colla Corii Asini(Ejiao). Zhongguo Zhong Yao Za Zhi 2020, 45, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Souroullas, K.; Aspri, M.; Papademas, P. Donkey milk as a supplement in infant formula: Benefits and technological challenges. Food Res. Int. 2018, 109, 416–425. [Google Scholar] [CrossRef]
- Carluccio, A.; Gloria, A.; Robbe, D.; Veronesi, M.C.; De Amicis, I.; Cairoli, F.; Contri, A. Reproductive characteristics of foal heat in female donkeys. Animal 2017, 11, 461–465. [Google Scholar] [CrossRef]
- Gougeon, A. Human ovarian follicular development: From activation of resting follicles to preovulatory maturation. Ann. D’endocrinol. 2010, 71, 132–143. [Google Scholar] [CrossRef]
- Magoffin, D.A. Ovarian theca cell. Int. J. Biochem. Cell Biol. 2005, 37, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Hsieh, M.; Zamah, A.M.; Oh, J.S. Novel signaling mechanisms in the ovary during oocyte maturation and ovulation. Mol. Cell. Endocrinol. 2012, 356, 65–73. [Google Scholar] [CrossRef]
- Gong, X.; Zhang, Y.; Ai, J.; Li, K. Application of Single-Cell RNA Sequencing in Ovarian Development. Biomolecules 2022, 13, 47. [Google Scholar] [CrossRef]
- Migliaccio, M.; Chioccarelli, T.; Ambrosino, C.; Suglia, A.; Manfrevola, F.; Carnevali, O.; Fasano, S.; Pierantoni, R.; Cobellis, G. Characterization of Follicular Atresia Responsive to BPA in Zebrafish by Morphometric Analysis of Follicular Stage Progression. Int. J. Endocrinol. 2018, 2018, 4298195. [Google Scholar] [CrossRef]
- Nicol, B.; Estermann, M.A.; Yao, H.H.; Mellouk, N. Becoming female: Ovarian differentiation from an evolutionary perspective. Front. Cell Dev. Biol. 2022, 10, 944776. [Google Scholar] [CrossRef] [PubMed]
- Schubert, B.; Canis, M.; Darcha, C.; Artonne, C.; Pouly, J.L.; Déchelotte, P.; Boucher, D.; Grizard, G. Human ovarian tissue from cortex surrounding benign cysts: A model to study ovarian tissue cryopreservation. Hum. Reprod. 2005, 20, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Ni, D.; Yang, H.; Zhao, F.; Yang, C.; Zhao, X.; Guo, Z.; Yu, K.; Wang, J.; Hu, Z.; et al. Deciphering the dynamics of the ovarian reserve in cynomolgus monkey through a quantitative morphometric study. Sci. Bull. 2022, 67, 1854–1859. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, X.; Zhu, K.; Yu, M.; Liu, Q.; De Felici, M.; Zhang, T.; Wang, J.; Shen, W. Sleep deprivation causes gut dysbiosis impacting on systemic metabolomics leading to premature ovarian insufficiency in adolescent mice. Theranostics 2024, 14, 3760–3776. [Google Scholar] [CrossRef]
- Lopes, K.R.F.; Lima, G.L.; Bezerra, L.G.P.; Barreto-Junior, R.A.; Oliveira, M.F.; Silva, A.R. Characterization of the ovarian preantral follicle populations and its correlation with age and nutritional status in Brazilian Northeastern donkeys (Equus assinus). Anim. Reprod. Sci. 2017, 187, 193–202. [Google Scholar] [CrossRef]
- Evans, A.C. Characteristics of ovarian follicle development in domestic animals. Reprod. Domest. Anim. 2003, 38, 240–246. [Google Scholar] [CrossRef]
- Pei, J.; Xiong, L.; Guo, S.; Wang, X.; Bao, P.; Wu, X.; Yan, P.; Guo, X. A single-cell transcriptomic atlas characterizes cell types and their molecular features in yak ovarian cortex. FASEB J. 2023, 37, e22718. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Tian, D.; Li, X.; Liu, S.; Tian, F.; Liu, D.; Wang, S.; Zhao, K. Multiomics Analyses Provide New Insight into Genetic Variation of Reproductive Adaptability in Tibetan Sheep. Mol. Biol. Evol. 2024, 41, msae058. [Google Scholar] [CrossRef]
- Liu, C.; Wu, T.; Fan, F.; Liu, Y.; Wu, L.; Junkin, M.; Wang, Z.; Yu, Y.; Wang, W.; Wei, W.; et al. A portable and cost-effective microfluidic system for massively parallel single-cell transcriptome profiling. bioRxiv 2019. [Google Scholar] [CrossRef]
- Lau, X.; Munusamy, P.; Ng, M.J.; Sangrithi, M. Single-Cell RNA Sequencing of the Cynomolgus Macaque Testis Reveals Conserved Transcriptional Profiles during Mammalian Spermatogenesis. Dev. Cell 2020, 54, 548–566.e7. [Google Scholar] [CrossRef]
- Fan, X.; Bialecka, M.; Moustakas, I.; Lam, E.; Torrens-Juaneda, V.; Borggreven, N.V.; Trouw, L.; Louwe, L.A.; Pilgram, G.S.K.; Mei, H.; et al. Single-cell reconstruction of follicular remodeling in the human adult ovary. Nat. Commun. 2019, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wei, X.; Liu, C.; Volpe, G.; Zhuang, Z.; Zou, X.; Wang, Z.; Pan, T.; Yuan, Y.; Zhang, X.; et al. Cell transcriptomic atlas of the non-human primate Macaca fascicularis. Nature 2022, 604, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, P.; Wang, J.; Ma, L.; E, W.; Suo, S.; Jiang, M.; Li, J.; Chen, H.; Sun, H.; et al. Construction of a cross-species cell landscape at single-cell level. Nucleic Acids Res. 2023, 51, 501–516. [Google Scholar] [CrossRef]
- Jiang, M.; Xiao, Y.; E, W.; Ma, L.; Wang, J.; Chen, H.; Gao, C.; Liao, Y.; Guo, Q.; Peng, J.; et al. Characterization of the Zebrafish Cell Landscape at Single-Cell Resolution. Front. Cell Dev. Biol. 2021, 9, 743421. [Google Scholar] [CrossRef]
- Fischer, S.; Crow, M.; Harris, B.D.; Gillis, J. Scaling up reproducible research for single-cell transcriptomics using MetaNeighbor. Nat. Protoc. 2021, 16, 4031–4067. [Google Scholar] [CrossRef]
- Van de Sande, B.; Flerin, C.; Davie, K.; De Waegeneer, M.; Hulselmans, G.; Aibar, S.; Seurinck, R.; Saelens, W.; Cannoodt, R.; Rouchon, Q.; et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 2020, 15, 2247–2276. [Google Scholar] [CrossRef] [PubMed]
- Suo, S.; Zhu, Q.; Saadatpour, A.; Fei, L.; Guo, G.; Yuan, G.C. Revealing the Critical Regulators of Cell Identity in the Mouse Cell Atlas. Cell Rep. 2018, 25, 1436–1445.e3. [Google Scholar] [CrossRef]
- Xiao, Z.; Dai, Z.; Locasale, J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019, 10, 3763. [Google Scholar] [CrossRef]
- Alghamdi, N.; Chang, W.; Dang, P.; Lu, X.; Wan, C.; Gampala, S.; Huang, Z.; Wang, J.; Ma, Q.; Zang, Y.; et al. A graph neural network model to estimate cell-wise metabolic flux using single-cell RNA-seq data. Genome Res. 2021, 31, 1867–1884. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Browaeys, R.; Saelens, W.; Saeys, Y. NicheNet: Modeling intercellular communication by linking ligands to target genes. Nat. Methods 2020, 17, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, J.S.; Han, X.; Ma, R.J.; Xu, J.L.; Xu, M.T.; Yuan, H.J.; Luo, M.J.; Tan, J.H. Expression profiling and function analysis identified new genes regulating cumulus expansion and apoptosis. Reproduction 2024, 168, e240128. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, S.; Shi, H.; Li, J.; Wan, X.; Sun, Y.; Li, H.; Cao, N.; Feng, Z.; Zhang, T.; et al. Revealing the Transcriptional and Metabolic Characteristics of Sebocytes Based on the Donkey Cell Transcriptome Atlas. Adv. Sci. 2025, 12, e2413819. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, Y.; Li, J.; Yu, Y.; Zhang, W.; Song, M.; Liu, Z.; Min, Z.; Hu, H.; Jing, Y.; et al. Single-Cell Transcriptomic Atlas of Primate Ovarian Aging. Cell 2020, 180, 585–600.e19. [Google Scholar] [CrossRef]
- Han, X.; Zhou, Z.; Fei, L.; Sun, H.; Wang, R.; Chen, Y.; Chen, H.; Wang, J.; Tang, H.; Ge, W.; et al. Construction of a human cell landscape at single-cell level. Nature 2020, 581, 303–309. [Google Scholar] [CrossRef]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Bunel, A.; Jorssen, E.P.; Merckx, E.; Leroy, J.L.; Bols, P.E.; Sirard, M.A. Individual bovine in vitro embryo production and cumulus cell transcriptomic analysis to distinguish cumulus-oocyte complexes with high or low developmental potential. Theriogenology 2015, 83, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Cui, Z.; Gao, Q.; Rui, R.; Xiong, B. Nicotinamide Mononucleotide Supplementation Reverses the Declining Quality of Maternally Aged Oocytes. Cell Rep. 2020, 32, 107987. [Google Scholar] [CrossRef]
- Plewes, M.R.; Krause, C.; Talbott, H.A.; Przygrodzka, E.; Wood, J.R.; Cupp, A.S.; Davis, J.S. Trafficking of cholesterol from lipid droplets to mitochondria in bovine luteal cells: Acute control of progesterone synthesis. FASEB J. 2020, 34, 10731–10750. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. Lipid droplets and steroidogenic cells. Exp. Cell Res 2016, 340, 209–214. [Google Scholar] [CrossRef]
- Martinez, C.A.; Rizos, D.; Rodriguez-Martinez, H.; Funahashi, H. Oocyte-cumulus cells crosstalk: New comparative insights. Theriogenology 2023, 205, 87–93. [Google Scholar] [CrossRef]
- Richards, J.S.; Ren, Y.A.; Candelaria, N.; Adams, J.E.; Rajkovic, A. Ovarian Follicular Theca Cell Recruitment, Differentiation, and Impact on Fertility: 2017 Update. Endocr. Rev. 2018, 39, 1–20. [Google Scholar] [CrossRef]
- Muhl, L.; Genove, G.; Leptidis, S.; Liu, J.; He, L.; Mocci, G.; Sun, Y.; Gustafsson, S.; Buyandelger, B.; Chivukula, I.V.; et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat. Commun. 2020, 11, 3953. [Google Scholar] [CrossRef]
- Paushter, D.H.; Du, H.; Feng, T.; Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018, 136, 1–17. [Google Scholar] [CrossRef]
- Dong, D.; Zhang, W.; Xiao, W.; Wu, Q.; Cao, Y.; Gao, X.; Huang, L.; Wang, Y.; Chen, J.; Wang, W.; et al. A GRN Autocrine-Dependent FAM135B/AKT/mTOR Feedforward Loop Promotes Esophageal Squamous Cell Carcinoma Progression. Cancer Res. 2021, 81, 910–922. [Google Scholar] [CrossRef]
- Seyiti, S.; Kelimu, A. Donkey Industry in China: Current Aspects, Suggestions and Future Challenges. J. Equine Vet. Sci. 2021, 102, 103642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, B.; Wang, Y.; Zhu, M.; Liu, G.; Wang, C. A survey report on the donkey original breeding farms in China: Current aspects and future prospective. Front. Vet. Sci. 2023, 10, 1126138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, T.; Zhang, Z.; Wang, C. Survival rate of donkey foals: Status quo and improvement methods. Afr. J. Biotechnol. 2021, 20, 340–348. [Google Scholar]
- Bocci, C.; Fanelli, D.; Moroni, R.; Tesi, M.; Camillo, F.; Rota, A.; Panzani, D. Pregnancy rates in Equids after Interspecific and Intraspecific Artificial Insemination. J. Equine Vet. Sci. 2022, 113, 103998. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Zhang, P.; Wang, R.; Mei, Y.; Sun, Z.; Fei, L.; Jiang, M.; Ma, L.; E, W.; et al. Deep learning of cross-species single-cell landscapes identifies conserved regulatory programs underlying cell types. Nat. Genet. 2022, 54, 1711–1720. [Google Scholar] [CrossRef]
- Vissenberg, R.; Manders, V.D.; Mastenbroek, S.; Fliers, E.; Afink, G.B.; Ris-Stalpers, C.; Goddijn, M.; Bisschop, P.H. Pathophysiological aspects of thyroid hormone disorders/thyroid peroxidase autoantibodies and reproduction. Hum. Reprod. Update 2015, 21, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yao, Y.; Ma, X.; Fu, B.; Li, N.; Zhang, C. Mechanisms of OCT4 on 3,5,3′-Tri-iodothyronine and FSH-induced Granulosa Cell Development in Female Mice. Endocrinology 2021, 162, bqab183. [Google Scholar] [CrossRef]
- Huang, J.C.; Duan, C.C.; Jin, S.; Sheng, C.B.; Wang, Y.S.; Yue, Z.P.; Guo, B. HB-EGF induces mitochondrial dysfunction via estrogen hypersecretion in granulosa cells dependent on cAMP-PKA-JNK/ERK-Ca(2+)-FOXO1 pathway. Int. J. Biol. Sci. 2022, 18, 2047–2059. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Q.; Hu, Y.; Xu, L.; Jiang, Y.; Zhang, C.; Ding, L.; Jiang, R.; Sun, J.; Sun, H.; et al. miR-181a increases FoxO1 acetylation and promotes granulosa cell apoptosis via SIRT1 downregulation. Cell Death Dis. 2017, 8, e3088. [Google Scholar] [CrossRef]
- Zhu, X.; Li, W.; Lu, M.; Shang, J.; Zhou, J.; Lin, L.; Liu, Y.; Xing, J.; Zhang, M.; Zhao, S.; et al. M(6)A demethylase FTO-stabilized exosomal circBRCA1 alleviates oxidative stress-induced granulosa cell damage via the miR-642a-5p/FOXO1 axis. J. Nanobiotechnol. 2024, 22, 367. [Google Scholar] [CrossRef]
- Sugiura, K.; Su, Y.Q.; Diaz, F.J.; Pangas, S.A.; Sharma, S.; Wigglesworth, K.; O’Brien, M.J.; Matzuk, M.M.; Shimasaki, S.; Eppig, J.J. Oocyte-derived BMP15 and FGFs cooperate to promote glycolysis in cumulus cells. Development 2007, 134, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Liu, H.; Guo, H.; Sun, L.; Gou, K.; Cui, S. RAF1 mediates the FSH signaling pathway as a downstream molecule to stimulate estradiol synthesis and secretion in mouse ovarian granulosa cells. Ann. Transl. Med. 2022, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Takahashi, N.; Azhary, J.M.; Kunitomi, C.; Fujii, T.; Osuga, Y. Endoplasmic reticulum stress: A key regulator of the follicular microenvironment in the ovary. Mol. Hum. Reprod. 2021, 27, gaaa088. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.M.; Duncan, W.C. Vascular morphogenesis in the primate ovary. Angiogenesis 2005, 8, 101–116. [Google Scholar] [CrossRef]
- Chen, J.; Fu, Y.; Day, D.S.; Sun, Y.; Wang, S.; Liang, X.; Gu, F.; Zhang, F.; Stevens, S.M.; Zhou, P.; et al. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat. Commun. 2017, 8, 383. [Google Scholar] [CrossRef]
- Wang, L.; Lin, L.; Qi, H.; Chen, J.; Grossfeld, P. Endothelial Loss of ETS1 Impairs Coronary Vascular Development and Leads to Ventricular Non-Compaction. Circ. Res. 2022, 131, 371–387. [Google Scholar] [CrossRef]
- Endo, T.; Kitajima, Y.; Nishikawa, A.; Manase, K.; Shibuya, M.; Kudo, R. Cyclic changes in expression of mRNA of vascular endothelial growth factor, its receptors Flt-1 and KDR/Flk-1, and Ets-1 in human corpora lutea. Fertil. Steril. 2001, 76, 762–768. [Google Scholar] [CrossRef]
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernandez-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis--brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782. [Google Scholar] [CrossRef]
- Liu, B.; Zhou, H.; Zhang, T.; Gao, X.; Tao, B.; Xing, H.; Zhuang, Z.; Dardik, A.; Kyriakides, T.R.; Goodwin, J.E. Loss of endothelial glucocorticoid receptor promotes angiogenesis via upregulation of Wnt/beta-catenin pathway. Angiogenesis 2021, 24, 631–645. [Google Scholar] [CrossRef]
- Gilchrist, R.B.; Lane, M.; Thompson, J.G. Oocyte-secreted factors: Regulators of cumulus cell function and oocyte quality. Hum. Reprod. Update 2008, 14, 159–177. [Google Scholar] [CrossRef]
- Aon, M.A.; Bernier, M.; Mitchell, S.J.; Di Germanio, C.; Mattison, J.A.; Ehrlich, M.R.; Colman, R.J.; Anderson, R.M.; de Cabo, R. Untangling Determinants of Enhanced Health and Lifespan through a Multi-omics Approach in Mice. Cell Metab. 2020, 32, 100–116 e104. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, W.; Ren, P.; Li, X.; Jin, J.; Dai, Y.; Pan, Y.; Jiang, L.; Fan, H.; Zhang, Y.; et al. Lysophosphatidic acid improves oocyte quality during IVM by activating the ERK1/2 pathway in cumulus cells and oocytes. Mol. Hum. Reprod. 2021, 27, gaab032. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.N.; Liu, X.L.; Li, N.; Zhang, R.Q.; Zhao, Y.; Feng, Y.Z.; Nyachoti, C.M.; Shen, W.; Li, L. Phosphatidylcholine could protect the defect of zearalenone exposure on follicular development and oocyte maturation. Aging 2018, 10, 3486–3506. [Google Scholar] [CrossRef]
- Zhang, C.H.; Liu, X.Y.; Wang, J. Essential Role of Granulosa Cell Glucose and Lipid Metabolism on Oocytes and the Potential Metabolic Imbalance in Polycystic Ovary Syndrome. Int. J. Mol. Sci. 2023, 24, 16247. [Google Scholar] [CrossRef]
- Cui, G.; Zhou, J.Y.; Ge, X.Y.; Sun, B.F.; Song, G.G.; Wang, X.; Wang, X.Z.; Zhang, R.; Wang, H.L.; Jing, Q.; et al. m(6) A promotes planarian regeneration. Cell Prolif. 2023, 56, e13481. [Google Scholar] [CrossRef]
- Dzafic, E.; Stimpfel, M.; Virant-Klun, I. Plasticity of granulosa cells: On the crossroad of stemness and transdifferentiation potential. J. Assist. Reprod. Genet. 2013, 30, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; McNeilly, A.S. Theca: The forgotten cell of the ovarian follicle. Reproduction 2010, 140, 489–504. [Google Scholar] [CrossRef]
- Shen, J.; Cao, B.; Wang, Y.; Ma, C.; Zeng, Z.; Liu, L.; Li, X.; Tao, D.; Gong, J.; Xie, D. Hippo component YAP promotes focal adhesion and tumour aggressiveness via transcriptionally activating THBS1/FAK signalling in breast cancer. J. Exp. Clin. Cancer Res. 2018, 37, 175. [Google Scholar] [CrossRef]
- Bauman, D.R.; Steckelbroeck, S.; Williams, M.V.; Peehl, D.M.; Penning, T.M. Identification of the major oxidative 3α-hydroxysteroid dehydrogenase in human prostate that converts 5α-androstane-3alpha,17β-diol to 5α-dihydrotestosterone: A potential therapeutic target for androgen-dependent disease. Mol. Endocrinol. 2006, 20, 444–458. [Google Scholar] [CrossRef]
- Wickenheisser, J.K.; Nelson-DeGrave, V.L.; Hendricks, K.L.; Legro, R.S.; Strauss, J.F., 3rd; McAllister, J.M. Retinoids and retinol differentially regulate steroid biosynthesis in ovarian theca cells isolated from normal cycling women and women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 4858–4865. [Google Scholar] [CrossRef]
- Gangestad, S.W.; Haselton, M.G. Human estrus: Implications for relationship science. Curr. Opin. Psychol. 2015, 1, 45–51. [Google Scholar] [CrossRef]
- Ajayi, A.F.; Akhigbe, R.E. Staging of the estrous cycle and induction of estrus in experimental rodents: An update. Fertil. Res. Pract. 2020, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- McGee, E.A.; Hsueh, A.J. Initial and cyclic recruitment of ovarian follicles. Endocr. Rev. 2000, 21, 200–214. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Niu, Y.; Zhang, X.; Wang, T.; Tian, Z.; Zhang, X.; Guo, J.; Ge, W.; Liu, S.; Sun, Y.; et al. The Transcriptomic Signature of Donkey Ovarian Tissue Revealed by Cross-Species Comparative Analysis at Single-Cell Resolution. Animals 2025, 15, 1761. https://doi.org/10.3390/ani15121761
Tian Y, Niu Y, Zhang X, Wang T, Tian Z, Zhang X, Guo J, Ge W, Liu S, Sun Y, et al. The Transcriptomic Signature of Donkey Ovarian Tissue Revealed by Cross-Species Comparative Analysis at Single-Cell Resolution. Animals. 2025; 15(12):1761. https://doi.org/10.3390/ani15121761
Chicago/Turabian StyleTian, Yu, Yilin Niu, Xinhao Zhang, Tao Wang, Zhe Tian, Xiaoyuan Zhang, Jiachen Guo, Wei Ge, Shuqin Liu, Yujiang Sun, and et al. 2025. "The Transcriptomic Signature of Donkey Ovarian Tissue Revealed by Cross-Species Comparative Analysis at Single-Cell Resolution" Animals 15, no. 12: 1761. https://doi.org/10.3390/ani15121761
APA StyleTian, Y., Niu, Y., Zhang, X., Wang, T., Tian, Z., Zhang, X., Guo, J., Ge, W., Liu, S., Sun, Y., Li, J., Shen, W., Wang, J., & Zhang, T. (2025). The Transcriptomic Signature of Donkey Ovarian Tissue Revealed by Cross-Species Comparative Analysis at Single-Cell Resolution. Animals, 15(12), 1761. https://doi.org/10.3390/ani15121761