
Enhancing Biosecurity in Mollusc Aquaculture: A Review of Current Isothermal Nucleic Acid Detection Methods

 ,
,  and
and 
Simple Summary
Abstract
1. Introduction
2. Pathogens of High Biosecurity Concern in Global Mollusc Production and Their Current Molecular Diagnostic Methods
2.1. Viral Diseases Affecting Molluscs
2.1.1. Abalone Viral Ganglioneuritis (AVG)
2.1.2. Abalone Shrivelling Syndrome (AbSS)
2.1.3. Acute Viral Necrobiotic Disease
2.1.4. Infection with Ostreid Herpesvirus-1
2.2. Parasitic Diseases Affecting Molluscs
2.2.1. Haplosporidiosis
2.2.2. Bonamiosis
2.2.3. Marteiliosis
2.2.4. Marteilioides
2.2.5. Denman Island Disease
2.2.6. Perkinsosis

{kind=link}
{kind=link}
{kind=link}
| Pathogen | Susceptible Mollusc (s) | Detection Method |
|---|---|---|
| Virus | ||
| Abalone herpesvirus (AbHV) | Blacklip abalone 1 Brown abalone 2 Disc abalone 2 Greenlip abalone 1 Pink abalone 2 Small abalone 1 Tiger abalone 1 | cPCR [131] Sequencing [131] qPCR [29,30] ISH [132] |
| Abalone shrivelling syndrome (ASSV) | Disc abalone Small abalone | qPCR [35] nPCR [34] |
| Acute Viral Necrobiotic Virus (AVNV) | Scallops | cPCR [41] qPCR [41,133,134] |
| Ostreid herpesvirus-1 (OsHV-1) | Ark clams Australian flat oyster Bay scallops Blood clam Blue mussels Chilean oyster European clam Flat oyster Great scallop Hairy mussels Manila clam Pacific oyster Portuguese oyster Sydney cockle Sydney rock oysters Telline Virescent oyster Whelks | PCR [48] ISH [51,52] qPCR [53] |
| Parasite | ||
| Bonamia spp. | Australian flat oyster 3 Chilean oyster 1 Crested oyster 1 Dwarf oyster 1 European flat oyster 1 Hawaiian oyster 1 Jinjiang oysters 2 Olympia oyster Pacific oyster 1 Portuguese oyster 1 Suminoe oyster 1 Sydney rock oysters | cPCR [77,78] qPCR [79] mPCR [18] ISH [78,80] |
| Bonamia exitiosa | Argentinian flat oyster Australian flat oyster 1 Chilean oyster 1 Dwarf oyster 1 Eastern oyster European flat oyster 1 Olympia oyster Pacific oyster Sydney rock oyster | qPCR [69] cPCR and sequencing [78,135,136] PCR-RFLP [72] mPCR [69] ISH [78,80,137] |
| Bonamia ostreae | Argentinian flat oyster 3 Asiatic oyster Australian flat oyster Chilean oyster European flat oyster 1 Pacific oyster 3 Portuguese oyster Suminoe oyster 1 | ISH [78,107] qPCR [79,138,139] cPCR [77,78,140] mPCR [69] PCR-RFLP [72] |
| Haplosporidium spp. | Australian flat oyster Blue mussel California mussel Cockles Eastern oyster European flat oyster Freshwater snails | qPCR [141,142] cPCR [60,143,144] mPCR [62] ISH [60] |
| Haplosporidium nelson | Eastern oyster Pacific oyster | ISH [145] cPCR [146] qPCR [147] mPCR [62] |
| Marteilia spp. | Argentinian flat oyster 3 Australian flat oyster Banded Carpet Shell Blacklip oyster Blacklip pearl oyster Blue mussel Calico scallop Chilean oyster Common cockle Dwarf oyster Eastern oyster European flat oyster Grooved razor clam Hooded oyster Iwagaki oyster Jackknife clam Manila clam Maxima clam Mediterranean mussel Northern horse mussel Pacific oyster Palourde clam Peppery furrow shell Pod razor Puelchean oyster Pullet carpet shell Rock oyster Striped venus clam Suminoe oyster Venerid clam | cPCR [89,90,93] ISH [93] RFLP-PCR [148] |
| Marteilia refringens | Argentinian flat oyster 2 Asiatic oyster 1 Australian flat oyster 2 Banded Carpet Shell Blue mussel 1 Calico scallop 2 Chilean oyster 1 Common cockle 1 Dwarf oyster 2 Eastern oyster 1 European flat oyster 1 Grooved razor clam 1 Hooded oyster 1 Jackknife clam Mediterranean mussel 1 Olympia oyster 1 Pacific oyster 2 Palourde clam Planktonic copepods 2 Pod razor Pullet carpet shell Small brown mussel 2 Striped venus clam 1 | nPCR [90,149] cPCR and sequencing [89,93,97] mPCR [18] qPCR [150] ISH [89,90,99,151] |
| Marteilia sydneyi | Flat oyster Sydney rock oyster | cPCR [152] mPCR [18] ISH [153] |
| Marteilioides spp. | Manila clam Northern blacklip oyster Pacific oyster Suminoe oyster | nPCR [154] |
| Marteilioides chungmuensis | Iwagaki oyster Manila clam Pacific oyster Pacific oyster Suminoe oyster | cPCR [102,155] ISH [102] |
| Mikrocytos mackini | Eastern oyster European flat oyster Olympia flat oyster Pacific oyster | cPCR [156] qPCR [108] ISH [157] FISH [156] |
| Perkinsus spp. | Asian littleneck clam Baltic clam Eastern oyster European flat oyster Hong Kong oyster Mangrove oyster Manila clam Palourde clam Soft shell clam Stout tagelus Suminoe oyster Sydney cockle 1 Yesso scallop | cPCR [122,158] ISH [124] PCR—DGGE 1 [159] mPCR-ELISA [129] |
| Perkinsus andrewsi | Baltic clam | cPCR [127] |
| Perkinsus atlanticus | Palourde clam | cPCR [160] mPCR-ELISA [129] |
| Perkinsosis marinus | Baltic macoma Blue mussel Cortez oyster 1 Eastern oyster 1 Mangrove oyster 1 Pacific oyster 1 Soft-shelled clam Suminoe oyster 1 | cPCR [123,158] ISH [124,126,161] qPCR [120,123] mPCR-ELISA [129] RFLP-PCR [162] |
| Perkinsosis olseni | Akoya pearl oyster 1 Asian littleneck clam 1 Australian flat oyster 1 Blacklip abalone 1 Blacklip pearl oyster 1 Crocus clam 1 European aurora venus clam 1 Giant clam 1 Greenlip abalone 1 Green-lipped mussel 1 Japanese pearl oyster 1 Kumamoto oyster Manila clam 1 Maxima clam 1 New Zealand ark shell 1 New Zealand cockle 1 New Zealand pauaa 1 New Zealand pipia 1 New Zealand scallop 1 Pacific oyster 1 Pearl oyster 1 Pullet carpet shell 1 Sand cockle Silverlip pearl oyster 1 Staircase abalone 1 Suminoe oyster 1 Sydney cockle 1 Venerid clam 1 Venerid commercial clam Venus clam Wedge shell Whirling abalone 1 | ISH [124,161,163] cPCR [123,161] qPCR [123] |
3. Isothermal Nucleic Acid Detection Methods
3.1. Loop Mediated Isothermal Amplification (LAMP)
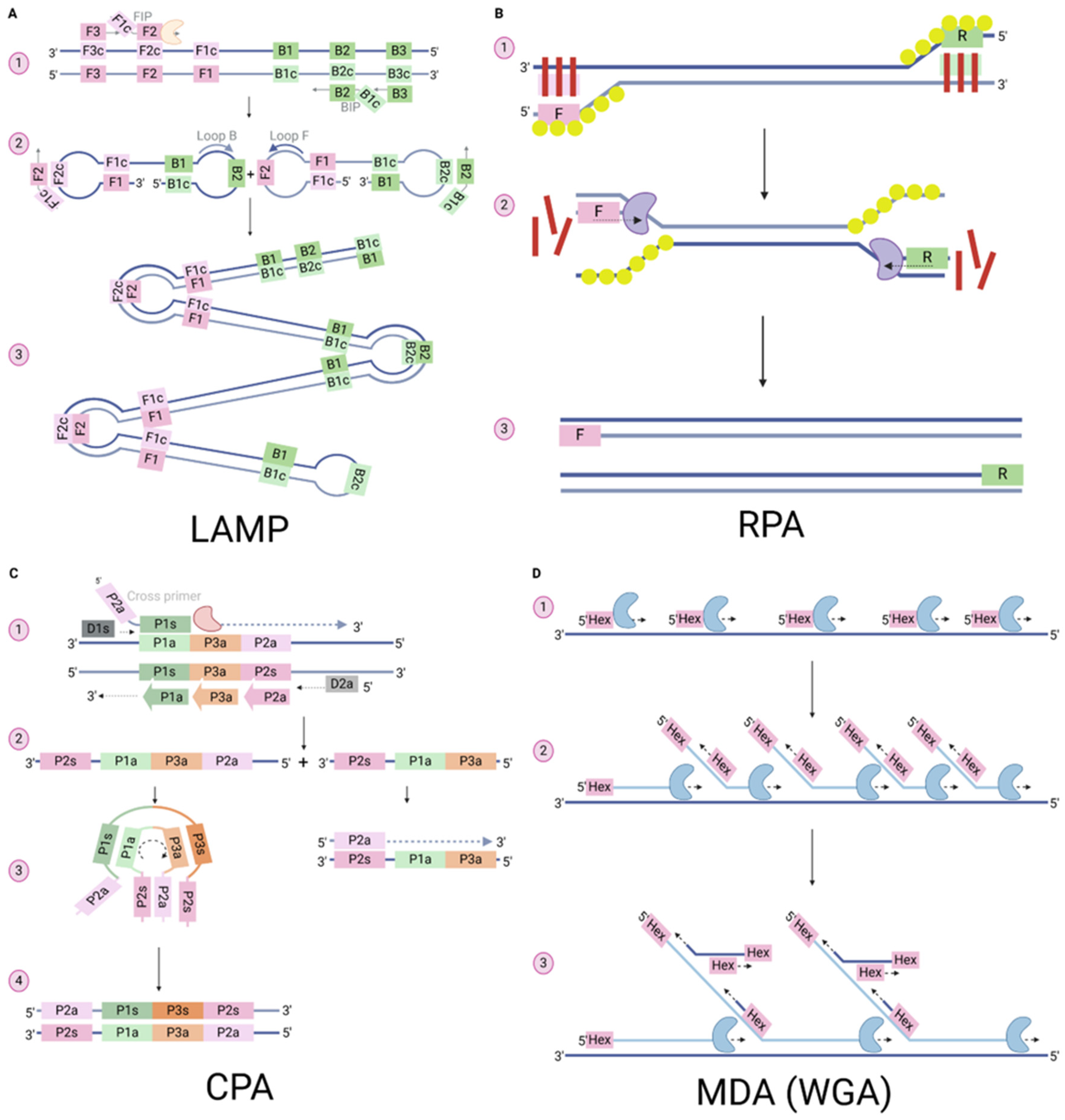
3.2. Recombinase Polymerase Amplification (RPA)
3.3. Cross-Priming Isothermal Amplification (CPA)
3.4. Multiple Displacement Amplification (MDA)
4. Application of Isothermal Amplification of Viral Pathogens Infecting Molluscs
5. Application of Isothermal Amplification of Parasitic Pathogens Infecting Molluscs
| Pathogen | Type | Target | Sample | Duration (minutes) | Sensitivity # | In-Field | Ref. |
|---|---|---|---|---|---|---|---|
| Virus | |||||||
| Abalone herpesvirus (AVG) | LAMP | DNA polymerase gene | Nerve tissues | 60 | 100 copies/µL | No | [176] |
| Abalone herpesvirus (AVG) | RPA | ORF38 | Muscle tissue | 20 | 100 copies | No | [170] |
| Acute Viral Necrobiotic Virus (AVNV) | LAMP | - | Tissues | 60 | 1 fg | No | [199] |
| Abalone shrivelling syndrome-associated virus (AbSV) | LAMP | ORF2 | Water | 60 | 10 copies | No | [200] |
| Ostreid herpesvirus (OsHV-1) | LAMP | ORF 109 | Tissues except for gonad and adductor muscle | 60 | 20 copies | No | [201] |
| Ostreid herpesvirus (OsHV-1) | LAMP | ORF 4 | Tissues | 60 | 103 copies | No | [202] |
| Ostreid herpesvirus (OsHV-1) | RPA | ORF 95 | Tissues | 20 | 207 copies | No | [204] |
| Ostreid herpesvirus (OsHV-1) | RPA | ORF 95 | Tissues | 20 | 5 copies | No | [203] |
| Ostreid herpesvirus (OsHV-1)-SB * | CPA | - | - | 60 | 30 copies/µL | No | [171] |
| Parasites | |||||||
| Bonamia exitiosa | MDA-WGA | Actin | Gill tissues | 90 | - | No | [19] |
| Bonamia exitiosa | LAMP | Actin | Gill tissues | 30 | 50 copies/µL | No | [206] |
| Bonamia ostreae | LAMP | Actin-1 | Gill tissues | 30 | 50 copies/µL | No | [206] |
| Bonamia spp. | LAMP | 18S | Gill tissues | 30 | 50 copies/µL | No | [206] |
| Marteilia refringens | LAMP | - | 60 | 20 fg | No | [207] | |
| Perkinsus spp. | LAMP | Internal transcribed spacer 2 (ITS-2) | Gills/body tissues | 49.8 | 10 copies of plasmid DNA | No | [177] |
| Perkinsus spp. | LAMP | ITS2 | Tissues | 30–60 | 3.6–36 ng | No | [208] |
| Perkinsus beihaiensis | RPA | ITS | Gills | 25 | 26 copies | No | [209] |
| Perkinsus olseni | LAMP | ITS 5.8S rDNA | - | 60 | 30 copies | No | [210] |
| Perkinsus olseni | LAMP | Between 5.8S and ITS 2 | - | - | 100 fg | No | [172] |
6. Future Improvements in the Application of Isothermal Amplification
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Worldometer. World Population. Available online: https://www.worldometers.info/world-population/ (accessed on 3 February 2025).
- Carnegie, R.B.; Arzul, I.; Bushek, D. Managing marine mollusc diseases in the context of regional and international commerce: Policy issues and emerging concerns. Phil. Trans. R. Soc. B 2016, 371, 20150215. [Google Scholar] [CrossRef] [PubMed]
- FAO. The State of World Fisheries and Aquaculture 2018-Meeting the Sustainable Development Goals; Fisheries and Aquaculture Department, Food and Agriculture Organization of the United Nations: Rome, Italy, 2020. [Google Scholar]
- FAO. The State of World Fisheries and Aquaculture 2024: Blue Transformation in Action. In The State of World Fisheries and Aquaculture (SOFIA); FAO: Rome, Italy, 2024. [Google Scholar]
- Benkendorff, K. Molluscan biological and chemical diversity: Secondary metabolites and medicinal resources produced by marine molluscs. Biol. Rev. 2010, 85, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Singh, S.; Pal, B. Superior adsorption removal of dye and high catalytic activity for transesterification reaction displayed by crystalline CaO nanocubes extracted from mollusc shells. Fuel Process. Technol. 2021, 213, 106707. [Google Scholar] [CrossRef]
- Marín Aguilera, B.; Iacono, F.; Gleba, M. Colouring the mediterranean: Production and consumption of purple-dyed textiles in pre-roman times. J. Mediterr. Archaeol. 2019, 31, 2. [Google Scholar] [CrossRef]
- Meng, X.; Yang, X.; Lin, G.; Fang, Y.; Ruan, Z.; Liu, M.; Liu, G.; Li, M.; Yang, D. Mannan oligosaccharide increases the growth performance, immunity and resistance capability against Vibro Parahemolyticus in juvenile abalone Haliotis discus hannai Ino. Fish. Shellfish. Immunol. 2019, 94, 654–660. [Google Scholar] [CrossRef]
- Arzul, I.; Corbeil, S.; Morga, B.; Renault, T. Viruses infecting marine molluscs. J. Invertebr. Pathol. 2017, 147, 118–135. [Google Scholar] [CrossRef]
- Renault, T.; Novoa, B. Viruses infecting bivalve molluscs. Aquat. Living Resour. 2004, 17, 397–409. [Google Scholar] [CrossRef]
- Paillard, C.; Le Roux, F.; Borrego, J.J. Bacterial disease in marine bivalves, a review of recent studies: Trends and evolution. Aquat. Living Resour. 2004, 17, 477–498. [Google Scholar] [CrossRef]
- Berthe, F.C.; Le Roux, F.; Adlard, R.D.; Figueras, A. Marteiliosis in molluscs: A review. Aquat. Living Resour. 2004, 17, 433–448. [Google Scholar] [CrossRef]
- Villalba, A.; Reece, K.S.; Ordás, M.C.; Casas, S.M.; Figueras, A. Perkinsosis in molluscs: A review. Aquat. Living Resour. 2004, 17, 411–432. [Google Scholar] [CrossRef]
- Gleason, F.H.; Gadd, G.M.; Pitt, J.I.; Larkum, A.W.D. The roles of endolithic fungi in bioerosion and disease in marine ecosystems. I. General concepts. Mycology 2017, 8, 205–215. [Google Scholar] [CrossRef]
- WOAH. Notifiable Animal Diseases. Available online: https://www.woah.org/en/what-we-do/animal-health-and-welfare/animal-diseases/?_tax_animal=aquatics%2Cmolluscs (accessed on 29 May 2025).
- Arzul, I.; Renault, T.; Lipart, C. Experimental herpes-like viral infections in marine bivalves: Demonstration of interspecies transmission. Dis. Aquat. Organ. 2001, 46, 1–6. [Google Scholar] [CrossRef]
- Arzul, I.; Renault, T.; Lipart, C.; Davison, A.J. Evidence for interspecies transmission of oyster herpesvirus in marine bivalves. J. Gen. Virol. 2001, 82, 865–870. [Google Scholar] [CrossRef]
- Canier, L.; Dubreuil, C.; Noyer, M.; Serpin, D.; Chollet, B.; Garcia, C.; Arzul, I. A new multiplex real-time PCR assay to improve the diagnosis of shellfish regulated parasites of the genus Marteilia and Bonamia. Prev. Vet. Med. 2020, 183, 105126. [Google Scholar] [CrossRef]
- Prado-Alvarez, M.; Couraleau, Y.; Chollet, B.; Tourbiez, D.; Arzul, I. Whole-genome amplification: A useful approach to characterize new genes in unculturable protozoan parasites such as Bonamia exitiosa. Parasitology 2015, 142, 1523–1534. [Google Scholar] [CrossRef]
- Bai, C.-M.; Li, Y.-N.; Chang, P.-H.; Jiang, J.-Z.; Xin, L.-S.; Li, C.; Wang, J.-Y.; Wang, C.-M. Susceptibility of two abalone species, Haliotis diversicolor supertexta and Haliotis discus hannai, to Haliotid herpesvirus 1 infection. J. Invertebr. Pathol. 2019, 160, 26–32. [Google Scholar] [CrossRef]
- Corbeil, S. Abalone Viral Ganglioneuritis. Pathogens 2020, 9, 720. [Google Scholar] [CrossRef]
- Chen, I.-W.; Chang, P.-H.; Chen, M.-S.; Renault, T.; Chen, M.-M.; Kuo, S.-T.; Cheng, C.-H. Exploring the chronic mortality affecting abalones in taiwan: Differentiation of abalone herpesvirus-associated acute infection from chronic mortality by pcr and in situ hybridization and histopathology. Taiwan Vet. J. 2016, 42, 1–9. [Google Scholar] [CrossRef]
- Mouton, A. An Epidemiological Study of Parasites Infecting the South African Abalone (Haliotis midae). In Western Cape Aquaculture Facilities; University of Pretoria (South Africa): Pretoria, South Africa, 2011. [Google Scholar]
- Chang, P.H.; Kuo, S.T.; Lai, S.H.; Yang, H.S.; Ting, Y.Y.; Hsu, C.L.; Chen, H.C. Herpes-like virus infection causing mortality of cultured abalone Haliotis diversicolor supertexta in Taiwan. Dis. Aquat. Organ. 2005, 65, 23–27. [Google Scholar] [CrossRef]
- Corbeil, S.; McColl, K.A.; Williams, L.M.; Mohammad, I.; Hyatt, A.D.; Crameri, S.G.; Fegan, M.; Crane, M.S. Abalone viral ganglioneuritis: Establishment and use of an experimental immersion challenge system for the study of abalone herpes virus infections in Australian abalone. Virus Res. 2012, 165, 207–213. [Google Scholar] [CrossRef]
- Corbeil, S.; McColl, K.A.; Williams, L.M.; Slater, J.; Crane, M.S.J. Innate resistance of New Zealand paua to abalone viral ganglioneuritis. J. Invertebr. Pathol. 2017, 146, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Z.X.; Feng, J.; Liu, G.F. Virus infection in cultured abalone, Haliotis diversicolor Reeve in Guangdong Province, China. J. Shellfish. Res. 2004, 23, 1163–1169. [Google Scholar]
- Hooper, C.; Hardy-Smith, P.; Handlinger, J. Ganglioneuritis causing high mortalities in farmed Australian abalone (Haliotis laevigata and Haliotis rubra). Aust. Vet. J. 2007, 85, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Caraguel, C.; Ellard, K.; Moody, N.; Corbeil, S.; Williams, L.; Mohr, P.; Cummins, D.; Hoad, J.; Slater, J.; Crane, M. Diagnostic test accuracy when screening for Haliotid herpesvirus 1 (AbHV) in apparently healthy populations of Australian abalone Haliotis spp. Dis. Aquat. Organ. 2019, 136, 199–207. [Google Scholar] [CrossRef]
- Corbeil, S.; Colling, A.; Williams, L.; Wong, F.; Savin, K.; Warner, S.; Murdoch, B.; Cogan, N.; Sawbridge, T.; Fegan, M.; et al. Development and validation of a TaqMan® PCR assay for the Australian abalone herpes-like virus. Dis. Aquat. Organ. 2010, 92, 1–10. [Google Scholar] [CrossRef]
- Chen, M.-H.; Chen, I.-W.; Kuo, S.-T.; Hsu, W.-L.; Chang, P.-H. Evaluation of a bacteriophage-related chimeric marine virus associated with abalone mortality in Taiwan. Taiwan Vet. J. 2014, 40, 77–82. [Google Scholar] [CrossRef]
- Wang, J. Spring explosive epidemic disease of abalone in Dongshan district. Xiamen Univ. Nat. Sci. 1999, 38, 644–654. [Google Scholar]
- Handlinger, J. General pathology and diseases of abalone. In Aquaculture Pathophysiology; Kibenge, F.S.B., Baldisserotto, B., Chong, R.S.-M., Eds.; Academic Press: New York, NY, USA, 2022; pp. 405–447. [Google Scholar]
- Jiang, J.-Z.; Liang, Y.-Y.; Luo, L.-J.; Guo, Z.-X.; Zhuang, J.; Liu, G.-F.; Su, Y.-L.; Wang, J.-Y. Nested PCR detection of abalone shriveling syndrome-associated virus in China. J. Virol. Methods 2012, 184, 21–26. [Google Scholar] [CrossRef]
- Jiang, J.-Z.; Zhu, Z.-N.; Zhang, H.; Liang, Y.-Y.; Guo, Z.-X.; Liu, G.-F.; Su, Y.-L.; Wang, J.-Y. Quantitative PCR detection for abalone shriveling syndrome-associated virus. J. Virol. Methods 2012, 184, 15–20. [Google Scholar] [CrossRef]
- Zhuang, J.; Cai, G.; Lin, Q.; Wu, Z.; Xie, L. A bacteriophage-related chimeric marine virus infecting abalone. PLoS ONE 2010, 5, e13850. [Google Scholar] [CrossRef]
- Yang, R. Molecular Epidemiological Investigation on Abalone Shriveling Syndrome-Associated Virus and Herpes-like Virus and Its Related Research; Shanghai Ocean University: Shanghai, China, 2013. [Google Scholar]
- Haixin, A. Artificial infection of cultured scallop Chlamys farreri by pathogen from acute virus necrobiotic disease. J. Fish. Sci. China 2003, 10, 386–391. [Google Scholar]
- Tang, B.; Liu, B.; Wang, X.; Yue, X.; Xiang, J. Physiological and immune responses of zhikong scallop Chlamys farreri to the acute viral necrobiotic virus infection. Fish. Shellfish. Immunol. 2010, 29, 42–48. [Google Scholar] [CrossRef]
- Chen, G.; Wang, C.; Zhang, C.; Wang, Y.; Xu, Z.; Wang, C. A preliminary study of differentially expressed genes of the scallop Chlamys farreri against acute viral necrobiotic virus (AVNV). Fish. Shellfish. Immunol. 2013, 34, 1619–1627. [Google Scholar] [CrossRef]
- Ren, W. Detection Methods, Sequence of the Complete Genome of Acute Viral Necrobiotic Virus Isolated from Scallop Chlamys farreri; Ocean University China: Qingdao, China, 2009. [Google Scholar]
- Wang, C.; Wang, X.; Song, X.; Huang, J.; Song, W. Purification and ultrastructure of a spherical virus in cultured scallop Chlamys farreri. Shuichan Xuebao 2002, 26, 180–183. [Google Scholar]
- Fu, C.; Song, W.; Li, Y. Monoclonal antibodies developed for detection of an epizootic virus associated with mass mortalities of cultured scallop Chlamys farreri. Dis. Aquat. Organ. 2005, 65, 17–22. [Google Scholar] [CrossRef]
- Li, Y.; He, G.; Wang, X.; Wang, C.; Song, W. Detection of Acute Virus Necrobiotic Disease Virus (AVND Virus) in Chlamys farreri Using ELISA Technique. Gaojishu Tongxun 2003, 13, 90–92. [Google Scholar]
- Fuhrmann, M. Ostreid herpesvirus disease. In Aquaculture Pathophysiology; Kibenge, F.S.B., Baldisserotto, B., Chong, R.S.-M., Eds.; Academic Press: New York, NY, USA, 2022; pp. 473–488. [Google Scholar]
- Burge, C.A.; Friedman, C.S.; Kachmar, M.L.; Humphrey, K.L.; Moore, J.D.; Elston, R.A. The first detection of a novel OsHV-1 microvariant in San Diego, California, USA. J. Invertebr. Pathol. 2021, 184, 107636. [Google Scholar] [CrossRef]
- Roque, A.; Carrasco, N.; Andree, K.; Lacuesta, B.; Elandaloussi, L.; Gairin, I.; Rodgers, C.; Furones, M.D. First report of OsHV-1 microvar in Pacific oyster (Crassostrea gigas) cultured in Spain. Aquaculture 2012, 324–325, 303–306. [Google Scholar] [CrossRef]
- Renault, T.; Lipart, C. Diagnosis of herpes-like virus infections in oysters using molecular techniques. In Aquaculture and Water: Fish Culture, Shellfish Culture and Water Usage; ResearchGate: Berlin, Germany, 1998. [Google Scholar]
- Schikorski, D.; Renault, T.; Saulnier, D.; Faury, N.; Moreau, P.; Pépin, J.-F. Experimental infection of Pacific oyster Crassostrea gigas spat by ostreid herpesvirus 1: Demonstration of oyster spat susceptibility. Vet. Res. 2011, 42, 27. [Google Scholar] [CrossRef]
- Arzul, I.; Renault, T.; Thébault, A.; Gérard, A. Detection of oyster herpesvirus DNA and proteins in asymptomatic Crassostrea gigas adults. Virus Res. 2002, 84, 151–160. [Google Scholar] [CrossRef]
- Barbosa-Solomieu, V.; Miossec, L.; Vázquez-Juárez, R.; Ascencio-Valle, F.; Renault, T. Diagnosis of Ostreid herpesvirus 1 in fixed paraffin-embedded archival samples using PCR and in situ hybridisation. J. Virol. Methods 2004, 119, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Lipart, C.; Renault, T. Herpes-like virus detection in infected Crassostrea gigas spat using DIG-labelled probes. J. Virol. Methods 2002, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pepin, J.F.; Riou, A.; Renault, T. Rapid and sensitive detection of ostreid herpesvirus 1 in oyster samples by real-time PCR. J. Virol. Methods 2008, 149, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Burreson, E.M.; Ford, S.E. A review of recent information on the Haplosporidia, with special reference to Haplosporidium nelsoni (MSX disease). Aquat. Living Resour. 2004, 17, 499–517. [Google Scholar] [CrossRef]
- Comps, M.; Pichot, Y. Fine spore structure of a haplosporidan parasitizing Crassostrea gigas: Taxonomic implications. Dis. Aquat. Org. 1991, 11, 73–77. [Google Scholar] [CrossRef]
- Andrews, J.D. Oyster Mortality Studies in Virginia IV. MSX in James River Public Seed Beds; Virginia Institute of Marine Science: Gloucester Point, VA, USA, 1964. [Google Scholar]
- Ford, S.E.; Haskin, H.H. History and epizootiology of Haplosporidium nelsoni (MSX), an oyster pathogen in Delaware Bay, 1957–1980. J. Invertebr. Pathol. 1982, 40, 118–141. [Google Scholar] [CrossRef]
- Ford, S.E.; Stokes, N.A.; Alcox, K.A.; Kraus, B.S.F.; Barber, R.D.; Carnegie, R.B.; Burreson, E.M. Investigating the life cycle of Haplosporidium nelsoni (MSX): A review. J. Shellfish. Res. 2018, 37, 679–693. [Google Scholar] [CrossRef]
- Barber, B.J.; Ford, S.E.; Littlewood, D. A physiological comparison of resistant and susceptible oysters Crassostrea virginica (Gmelin) exposed to the endoparasite Haplosporidium nelsoni (Haskin, Stauber & Mackin). J. Exp. Mar. Biol. Ecol. 1991, 146, 101–112. [Google Scholar] [CrossRef]
- Stokes, N.A.; Burreson, E.M. Differential diagnosis of mixed Haplosporidium costale and Haplosporidium nelsoni infections in the eastern oyster, Crassostrea virginica, using DNA probes. J. Shellfish. Res. 2001, 20, 207. [Google Scholar]
- Arzul, I.; Carnegie, R.B. New perspective on the Haplosporidian parasites of molluscs. J. Invertebr. Pathol. 2015, 131, 32–42. [Google Scholar] [CrossRef]
- Penna, M.S.; Khan, M.; French, R.A. Development of a multiplex PCR for the detection of Haplosporidium nelsoni, Haplosporidium costale and Perkinsus marinus in the eastern oyster (Crassostrea virginica, Gmelin, 1971). Mol. Cell. Probes 2001, 15, 385–390. [Google Scholar] [CrossRef]
- Carnegie, R.B.; Burreson, E.M.; Hine, P.M.; Stokes, N.A.; Audemard, C.; Bishop, M.J.; Peterson, C.H. Bonamia perspora n. sp. (Haplosporidia), a parasite of the oyster Ostreola equestris, is the first Bonamia species known to produce spores. J. Eukaryot. Microbiol. 2006, 53, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Dinamani, P.; Hine, P.; Jones, J. Occurrence and characteristics of the haemocyte parasite Bonamia ostreae in the New Zealand dredge oyster Tiostrea lutaria. Dis. Aquat. Organ. 1987, 3, 37–44. [Google Scholar] [CrossRef]
- Culloty, S.C.; Mulcahy, M.F. Bonamia ostreae in the native oyster Ostrea edulis. Fish. Bull. Mar. Inst. Dublin. 2007. Available online: https://oar.marine.ie/handle/10793/269 (accessed on 29 May 2025).
- European Commission. Commission implementing regulations (EU) 2018/1882. In Official Journal of the European Union; European Commission: Brussels, Belgium, 2022. [Google Scholar]
- Bower, S.M.; McGladdery, S.E.; Price, I.M. Synopsis of infectious diseases and parasites of commercially exploited shellfish. Annu. Rev. Fish. Dis. 1994, 4, 1–199. [Google Scholar] [CrossRef]
- Jørgensen, L.v.G.; Nielsen, J.W.; Villadsen, M.K.; Vismann, B.; Dalvin, S.; Mathiessen, H.; Madsen, L.; Kania, P.W.; Buchmann, K. A non-lethal method for detection of Bonamia ostreae in flat oyster (Ostrea edulis) using environmental DNA. Sci. Rep. 2020, 10, 16143. [Google Scholar] [CrossRef]
- Ramilo, A.; Navas, J.; Villalba, A.; Abollo, E. Species-specific diagnostic assays for Bonamia ostreae and B. exitiosa in European flat oyster Ostrea edulis: Conventional, real-time and multiplex PCR. Dis. Aquat. Organ. 2013, 104, 149–161. [Google Scholar] [CrossRef]
- Pichot, Y.; Comps, M.; Tige, G.; Grizel, H. Recherches sur Bonamia ostreae gen. n., sp. n., parasite nouveau de l’huître plate Ostrea edulis L. Rev. Trav. Off. Pech. Marit. 1979, 43, 131–140. [Google Scholar]
- Van Banning, P. The life cycle of the oyster pathogen Bonamia ostreae with a presumptive phase in the ovarian tissue of the European flat oyster, Ostrea edulis. Aquaculture 1990, 84, 189–192. [Google Scholar] [CrossRef]
- Abollo, E.; Ramilo, A.; Casas, S.M.; Comesaña, P.; Cao, A.; Carballal, M.J.; Villalba, A. First detection of the protozoan parasite Bonamia exitiosa (Haplosporidia) infecting flat oyster Ostrea edulis grown in European waters. Aquaculture 2008, 274, 201–207. [Google Scholar] [CrossRef]
- Lane, H.S. Studies on Bonamia parasites (Haplosporidia) in the New Zealand Flat Oyster Ostrea chilensis; University of Otago: Dunedin, New Zealand, 2018. [Google Scholar]
- Hine, P.; Bower, S.; Meyer, G.; Cochennec-Laureau, N.; Berthe, F. Ultrastructure of Mikrocytos mackini, the cause of Denman Island disease in oysters Crassostrea spp. and Ostrea spp. in British Columbia, Canada. Dis. Aquat. Organ. 2001, 45, 215–227. [Google Scholar] [CrossRef]
- Culloty, S.C.; Cronin, M.A.; Mulcahy, M.F. Potential resistance of a number of populations of the oyster Ostrea edulis to the parasite Bonamia ostreae. Aquaculture 2004, 237, 41–58. [Google Scholar] [CrossRef]
- Van Banning, P. Observations on bonamiasis in the stock of the European flat oyster, Ostrea edulis, in the Netherlands, with special reference to the recent developments in Lake Grevelingen. Aquaculture 1991, 93, 205–211. [Google Scholar] [CrossRef]
- Carnegie, R.B.; Barber, B.J.; Culloty, S.C.; Figueras, A.J.; Distel, D.L. Development of a PCR assay for detection of the oyster pathogen Bonamia ostreae and support for its inclusion in the Haplosporidia. Dis. Aquat. Organ. 2000, 42, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Cochennec, N.; Le Roux, F.; Berthe, F.; Gerard, A. Detection of Bonamia ostreae Based on Small Subunit Ribosomal Probe. J. Invertebr. Pathol. 2000, 76, 26–32. [Google Scholar] [CrossRef]
- Corbeil, S.; Arzul, I.; Diggles, B.; Heasman, M.; Chollet, B.; Berthe, F.; Crane, M. Development of a TaqMan PCR assay for the detection of Bonamia species. Dis. Aquat. Organ. 2006, 71, 75–80. [Google Scholar] [CrossRef]
- Hill, K.M.; Carnegie, R.B.; Aloui-Bejaoui, N.; El Gharsalli, R.; White, D.M.; Stokes, N.A.; Burreson, E.M. Observation of a Bonamia sp. infecting the oyster Ostrea stentina in Tunisia, and a consideration of its phylogenetic affinities. J. Invertebr. Pathol. 2010, 103, 179–185. [Google Scholar] [CrossRef]
- Carrasco, N.; Villalba, A.; Andree, K.; Engelsma, M.; Lacuesta, B.; Ramilo, A.; Gairín, I.; Furones, M.D. Bonamia exitiosa (Haplosporidia) observed infecting the European flat oyster Ostrea edulis cultured on the Spanish Mediterranean coast. J. Invertebr. Pathol. 2012, 110, 307–313. [Google Scholar] [CrossRef]
- Hine, P. Severe apicomplexan infection in the oyster Ostrea chilensis: A possible predisposing factor in bonamiosis. Dis. Aquat. Organ. 2002, 51, 49–60. [Google Scholar] [CrossRef]
- Calvo, L.M.R.; Calvo, G.W.; Burreson, E.M. Dual disease resistance in a selectively bred eastern oyster, Crassostrea virginica, strain tested in Chesapeake Bay. Aquaculture 2003, 220, 69–87. [Google Scholar] [CrossRef]
- Comps, M. Marteilia lengehi n. sp., parasite de l’huître Crassostrea cucullata Born. Rev. Trav. Off. Pech. Marit. 1976, 40, 347–349. [Google Scholar]
- Perkins, F.O.; Wolf, P.H. Fine structure of Marteilia sydneyi sp. n.: Haplosporidan pathogen of Australian oysters. J. Parasitol. 1976, 62, 528–538. [Google Scholar] [CrossRef]
- Villalba, A.; Iglesias, D.; Ramilo, A.; Darriba, S.; Parada, J.; No, E.; Abollo, E.; Molares, J.; Carballal, M. Cockle Cerastoderma edule fishery collapse in the Ría de Arousa (Galicia, NW Spain) associated with the protistan parasite Marteilia cochillia. Dis. Aquat. Organ. 2014, 109, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Norton, J.; Perkins, F.; Ledua, E. Marteilia-like infection in a giant clam, Tridacna maxima, in Fiji. J. Invertebr. Pathol. 1993, 61, 328–330. [Google Scholar] [CrossRef]
- Carrasco, N.; Green, T.; Itoh, N. Marteilia spp. parasites in bivalves: A revision of recent studies. J. Invertebr. Pathol. 2015, 131, 43–57. [Google Scholar] [CrossRef]
- Le Roux, F.; Audemard, C.; Barnaud, A.; Berthe, F. DNA probes as potential tools for the detection of Marteilia refringens. Mar. Biotechnol. 1999, 1, 588–597. [Google Scholar] [CrossRef]
- López-Flores, I.; de la Herrán, R.; Garrido-Ramos, M.A.; Navas, J.I.; Ruiz-Rejón, C.; Ruiz-Rejón, M. The molecular diagnosis of Marteilia refringens and differentiation between Marteilia strains infecting oysters and mussels based on the rDNA IGS sequence. Parasitology 2004, 129, 411–419. [Google Scholar] [CrossRef]
- Birch, G.; Apostolatos, C.; Taylor, S. A remarkable recovery in the Sydney rock oyster (Saccostrea glomerata) population in a highly urbanised estuary (Sydney estuary, Australia). J. Coast. Res. 2013, 29, 1009–1015. [Google Scholar] [CrossRef]
- Grizel, H. Etude des Récentes Épizooties de L’huître Plate Ostrea edulis Linné et de leur Impact sur L’ostréiculture Bretonne; Universite des Sciences et Techniques du Languedoc: Montpellier, France, 1985. [Google Scholar]
- LE Roux, F.; Lorenzo, G.; Peyret, P.; Audemard, C.; Figueras, A.; Vivarès, C.; Gouy, M.; Berthe, F. Molecular evidence for the existence of two species of Marteilia in Europe. J. Eukaryot. Microbiol. 2001, 48, 449–454. [Google Scholar] [CrossRef]
- Audemard, C.; LE Roux, F.; Barnaud, A.; Collins, C.; Sautour, B.; Sauriau, P.-G.; DE Montaudouin, X.; Coustau, C.; Combes, C.; Berthe, F. Needle in a haystack: Involvement of the copepod Paracartia grani in the life-cycle of the oyster pathogen Marteilia refringens. Parasitology 2002, 124, 315–323. [Google Scholar] [CrossRef]
- Villalba, A.; Mourelle, S.; López, M.; Carballal, M.; Azevedo, C. Marteiliasis affecting cultured mussels Mytilus galloprovincialis of Galicia (NW Spain). I. Etiology, phases of the infection, and temporal and spatial variability in prevalence. Dis. Aquat. Organ. 1993, 16, 61–72. [Google Scholar] [CrossRef]
- Wetchateng, T.; Friedman, C.; Wight, N.; Lee, P.; Teng, P.; Sriurairattana, S.; Wongprasert, K.; Withyachumnarnkul, B. Withering syndrome in the abalone Haliotis diversicolor supertexta. Dis. Aquat. Organ. 2010, 90, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Grizel, H.; Comps, M.; Cousserans, F.; Duthoit, J.L.; Le Pennec, M.A. Recherche sur l’agent de la maladie de la glande digestive de Ostrea edulis Linné. Sci. Pech. 1974, 7–30. [Google Scholar]
- Grizel, H.; Comps, M.; Cousserans, F.; Jean-robert, B.; Constantin, V. Etude d’un parasite de la glande digestive observé au cours de l’épizootie actuelle de l’huître plate. C. R. Acad. Sci. 1974, 279, 783–785. [Google Scholar]
- Thébault, A.; Bergman, S.; Pouillot, R.; Le Roux, F.; Berthe, F. Validation of in situ hybridisation and histology assays for the detection of the oyster parasite Marteilia refringens. Dis. Aquat. Organ. 2005, 65, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.K. Preliminary studies on the sporozoan parasites in oysters on the southern coast of Korea. Bull. Korean Fish. Soc. 1972, 5, 76–82. [Google Scholar]
- Comps, M.; Desportes, I. Etude ultrastructurale des Marteilioides chungmuensis NG, N. SP. parasite des ovocytes de l’huître Crassostrea gigas Th. Protistologica 1986, 22, 279–285. [Google Scholar]
- Itoh, N.; Oda, T.; Yoshinaga, T.; Ogawa, K. DNA probes for detection of Marteilioides chungmuensis from the ovary of Pacific oyster Crassostrea gigas. Fish. Pathol. 2003, 38, 163–169. [Google Scholar] [CrossRef]
- Ngo, T.; Berthe, F.; Choi, K. Prevalence and infection intensity of the ovarian parasite Marteilioides chungmuensis during an annual reproductive cycle of the oyster Crassostrea gigas. Dis. Aquat. Organ. 2003, 56, 259–267. [Google Scholar] [CrossRef]
- Anderson, T.J.; Lester, R.J.G. Sporulation of Marteilioides branchialis n. sp. (Paramyxea) in the Sydney rock oyster, Saccostrea commercialis: An electron microscope study. J. Protozool. 1992, 39, 502–508. [Google Scholar] [CrossRef]
- Choi, H.J.; Hwang, J.Y.; Choi, D.L.; Huh, M.D.; Park, M.A. A study of diagnostic methods for Marteilioides chungmuensis infections in the Pacific oyster Crassostrea gigas. J. Invertebr. Pathol. 2012, 111, 27–32. [Google Scholar] [CrossRef]
- Burki, F.; Corradi, N.; Sierra, R.; Pawlowski, J.; Meyer, G.R.; Abbott, C.L.; Keeling, P.J. Phylogenomics of the Intracellular Parasite Mikrocytos mackini Reveals Evidence for a Mitosome in Rhizaria. Curr. Biol. 2013, 23, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Carnegie, R.B.; Barber, B.J.; Distel, D.L. Detection of the oyster parasite Bonamia ostreae by fluorescent in situ hybridization. Dis. Aquat. Organ. 2003, 55, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Polinski, M.; Lowe, G.; Meyer, G.; Corbeil, S.; Colling, A.; Caraguel, C.; Abbott, C.L. Molecular detection of Mikrocytos mackini in Pacific oysters using quantitative PCR. Mol. Biochem. Parasitol. 2015, 200, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Auzoux-Bordenave, S. In vitro sporulation of the clam pathogen Perkinsus atlanticus (Apicomplexa, Perkinsea) under various environmental conditions. Oceanogr. Lit. Rev. 1996, 9, 926. [Google Scholar]
- Park, K.-I.; Choi, K.-S. Application of enzyme-linked immunosorbent assay for studying of reproduction in the Manila clam Ruditapes philippinarum (Mollusca: Bivalvia): I. Quantifying eggs. Aquaculture 2004, 241, 667–687. [Google Scholar] [CrossRef]
- Park, K.-I.; Tsutsumi, H.; Hong, J.-S.; Choi, K.-S. Pathology survey of the short-neck clam Ruditapes philippinarum occurring on sandy tidal flats along the coast of Ariake Bay, Kyushu, Japan. J. Invertebr. Pathol. 2008, 99, 212–219. [Google Scholar] [CrossRef]
- Neto, M.P.D.; Gesteira, T.C.V.; Sabry, R.C.; Feijó, R.G.; Forte, J.M.; Boehs, G.; Maggioni, R. First record of Perkinsus chesapeaki infecting Crassostrea rhizophorae in South America. J. Invertebr. Pathol. 2016, 141, 53–56. [Google Scholar] [CrossRef]
- Hanrio, E.; Batley, J.; Dungan, C.; Dang, C. Immunoassays and diagnostic antibodies for Perkinsus spp. pathogens of marine molluscs. Dis. Aquat. Organ. 2021, 147, 13–23. [Google Scholar] [CrossRef]
- Dungan, C.F.; Reece, K.S. 5.2.1 Perkinsus spp. Infections of Marine Molluscs. AFS-FHS. 2020. Available online: https://units.fisheries.org/fhs/wp-content/uploads/sites/30/2021/03/5.2.1-Perkinsus-2021.pdf (accessed on 29 May 2025).
- Valencia, J.; Bassitta, M.; Picornell, A.; Ramon, C.; Castro, J. New data on Perkinsus mediterraneus in the Balearic Archipelago: Locations and affected species. Dis. Aquat. Organ. 2014, 112, 69–82. [Google Scholar] [CrossRef]
- Andrews, J.D. Epizootiology of the Disease Caused by the Oyster Pathogen Perkinsus marinus and Its Effects on the Oyster Industry; American Fisheries Society: Washington, DC, USA, 1988; p. 18. [Google Scholar]
- Cook, T.; Folli, M.; Klinck, J.; Ford, S.; Miller, J. The Relationship Between Increasing Sea-Surface Temperature and the Northward Spread of Perkinsus marinus (Dermo) Disease Epizootics in Oysters. Estuar. Coast. Shelf Sci. 1998, 46, 587–597. [Google Scholar] [CrossRef]
- Pires, D.; Grade, A.; Ruano, F.; Afonso, F. Histopathologic Lesions in Bivalve Mollusks Found in Portugal: Etiology and Risk Factors. J. Mar. Sci. Eng. 2022, 10, 133. [Google Scholar] [CrossRef]
- Casas, S.M.; Villalba, A.; Reece, K.S. Study of perkinsosis in the carpet shell clam Tapes decussatus in Galicia (NW Spain). I. Identification of the aetiological agent and in vitro modulation of zoosporulation by temperature and salinity. Dis. Aquat. Organ. 2002, 50, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.D.; Miller, C.R.; Wilbur, A.E. TaqMan® MGB real-time PCR approach to quantification of Perkinsus marinus and Perkinsus spp. in oysters. J. Shellfish. Res. 2006, 25, 619–624. [Google Scholar] [CrossRef]
- Lenaers, G.; Maroteaux, L.; Michot, B.; Herzog, M. Dinoflagellates in evolution. A molecular phylogenetic analysis of large subunit ribosomal RNA. J. Mol. Evol. 1989, 29, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.A.; Xiao, J.; Dungan, C.F.; Reece, K.S. Description of Perkinsus beihaiensis n. sp., a new Perkinsus sp. parasite in oysters of southern China. J. Eukaryot. Microbiol. 2008, 55, 117–130. [Google Scholar] [CrossRef]
- Audemard, C.; Reece, K.S.; Burreson, E.M. Real-time PCR for detection and quantification of the protistan parasite Perkinsus marinus in environmental waters. Appl. Environ. Microbiol. 2004, 70, 6611–6618. [Google Scholar] [CrossRef]
- Elston, R.A.; Dungan, C.F.; Meyers, T.R.; Reece, K.S. Perkiasus sp infection risk for manila clams, Venerupis philippinarum (A. Adams and Reeve, 1850) on the Pacific coast of North and Central America. J. Shellfish. Res. 2004, 23, 101. [Google Scholar]
- Ramilo, A.; Carrasco, N.; Reece, K.S.; Valencia, J.M.; Grau, A.; Aceituno, P.; Rojas, M.; Gairin, I.; Furones, M.D.; Abollo, E.; et al. Update of information on perkinsosis in NW Mediterranean coast: Identification of Perkinsus spp. (Protista) in new locations and hosts. J. Invertebr. Pathol. 2015, 125, 37–41. [Google Scholar] [CrossRef]
- Reece, K.S.; Dungan, C.F.; Burreson, E.M. Molecular epizootiology of Perkinsus marinus and P. chesapeaki infections among wild oysters and clams in Chesapeake Bay, USA. Dis. Aquat. Organ. 2008, 82, 237–248. [Google Scholar] [CrossRef]
- Coss, C.A.; Robledo, J.A.F.; Ruiz, G.M.; Vasta, G.R. Description of Perkinsus andrewsi n. sp. isolated from the Baltic clam (Macoma balthica) by characterization of the ribosomal RNA locus, and development of a species-specific PCR-based diagnostic assay. J. Eukaryot. Microbiol. 2001, 48, 52–61. [Google Scholar] [CrossRef]
- Kang, H.-S.; Yang, H.-S.; Reece, K.S.; Cho, Y.-G.; Lee, H.-M.; Kim, C.-W.; Choi, K.-S. Survey on Perkinsus species in Manila clam Ruditapes philippinarum in Korean waters using species-specific PCR. Fish. Pathol. 2017, 52, 202–205. [Google Scholar] [CrossRef]
- Elandalloussi, L.M.; Leite, R.M.; Afonso, R.; Nunes, P.A.; Robledo, J.A.; Vasta, G.R.; Cancela, M. Development of a PCR-ELISA assay for diagnosis of Perkinsus marinus and Perkinsus atlanticus infections in bivalve molluscs. MCP 2004, 18, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.A. Characterization of exotic pathogens associated with the suminoe oyster, Crassostrea ariakensis. 2007. Available online: https://scholarworks.wm.edu/etd/1539616784/ (accessed on 29 May 2025).
- Chen, M.; Kuo, S.; Renault, T.; Friedman, C.; Chang, P. Development of a polymerase chain reaction for the detection of abalone herpesvirus infection based on the DNA polymerase gene. J. Virol. Methods 2012, 185, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fegan, M.; Kvalheim, N.; Wong, F.; Mohammad, I.; Savin, K.; Lancaster, M.; Crane, M.S.J.; Warner, M.S. Development of an in situ hybridisation assay for the detection and identification of the abalone herpes-like virus. In Proceedings of the 4th FRDC Aquatic Animal Health Subprogram Scientific Conference, Cairns, Australia, 22–24 July 2009. [Google Scholar]
- Ren, W.; Wang, C.; Sun, S.; Cai, Y.; Li, Y.; Yu, Z. Development and application of a FQ-PCR assay for detection of Chlamys farreri acute viral necrobiotic virus. JFSC 2009, 16, 564–571. [Google Scholar]
- Xing, J.; Lin, T.; Zhan, W. Variations of enzyme activities in the haemocytes of scallop Chlamys farreri after infection with the acute virus necrobiotic virus (AVNV). Fish. Shellfish. Immunol. 2008, 25, 847–852. [Google Scholar] [CrossRef]
- Narcisi, V.; Arzul, I.; Cargini, D.; Mosca, F.; Calzetta, A.; Traversa, D.; Robert, M.; Joly, J.P.; Chollet, B.; Renault, T.; et al. Detection of Bonamia ostreae and B. exitiosa (Haplosporidia) in Ostrea edulis from the Adriatic Sea (Italy). Dis. Aquat. Organ. 2010, 89, 79–85. [Google Scholar] [CrossRef]
- Carnegie, R.B.; Cochennec-Laureau, N. Microcell parasites of oysters: Recent insights and future trends. Aquat. Living Resour. 2004, 17, 519–528. [Google Scholar] [CrossRef]
- Ramilo, A.; Villalba, A.; Abollo, E. Species-specific oligonucleotide probe for detection of Bonamia exitiosa (Haplosporidia) using in situ hybridisation assay. Dis. Aquat. Organ. 2014, 110, 81–91. [Google Scholar] [CrossRef]
- Marty, G.D.; Bower, S.M.; Clarke, K.R.; Meyer, G.; Lowe, G.; Osborn, A.L.; Chow, E.P.; Hannah, H.; Byrne, S.; Sojonky, K.; et al. Histopathology and a real-time PCR assay for detection of Bonamia ostreae in Ostrea edulis cultured in western Canada. Aquaculture 2006, 261, 33–42. [Google Scholar] [CrossRef]
- Robert, M.; Garcia, C.; Chollet, B.; Lopez-Flores, I.; Ferrand, S.; François, C.; Joly, J.-P.; Arzul, I. Molecular detection and quantification of the protozoan Bonamia ostreae in the flat oyster, Ostrea edulis. Mol. Cell. Probes 2009, 23, 264–271. [Google Scholar] [CrossRef]
- Engelsma, M.; Kerkhoff, S.; Roozenburg, I.; Haenen, O.; van Gool, A.; Sistermans, W.; Wijnhoven, S.; Hummel, H. Epidemiology of Bonamia ostreae infecting european flat oysters Ostrea edulis from Lake Grevelingen, The Netherlands. Mar. Ecol. Prog. Ser. 2010, 409, 131–142. [Google Scholar] [CrossRef]
- Xie, Z.; Xie, L.; Fan, Q.; Pang, Y.; Deng, X.; Xie, Z.Q.; Liu, J.; Khan, M.I. A duplex quantitative real-time PCR assay for the detection of Haplosporidium and Perkinsus species in shellfish. Parasitol. Res. 2013, 112, 1597–1606. [Google Scholar] [CrossRef]
- Arzul, I.; Garcia, C.; Chollet, B.; Serpin, D.; Lupo, C.; Noyer, M.; Tourbiez, D.; Berland, C.; Dégremont, L.; Travers, M. First characterization of the parasite Haplosporidium costale in France and development of a real-time PCR assay for its rapid detection in the Pacific oyster, Crassostrea gigas. Transbound. Emerg. Dis. 2022, 69, e2041–e2058. [Google Scholar] [CrossRef]
- Lopez-Nuñez, R.; Melendreras, E.C.; Casalduero, F.G.; Prado, P.; Lopez-Moya, F.; Lopez-Llorca, L.V. Detection of Haplosporidium pinnae from Pinna nobilis Faeces. J. Mar. Sci. Eng. 2022, 10, 276. [Google Scholar] [CrossRef]
- Carella, F.; Antuofermo, E.; Farina, S.; Salati, F.; Mandas, D.; Prado, P.; Panarese, R.; Marino, F.; Fiocchi, E.; Pretto, T.; et al. In the wake of the ongoing mass mortality events: Co-occurrence of Mycobacterium, Haplosporidium and other pathogens in Pinna nobilis collected in Italy and Spain (Mediterranean Sea). Front. Mar. Sci. 2020, 7, 48. [Google Scholar] [CrossRef]
- Stokes, N.A.; Burreson, E.M. A sensitive and specific DNA probe for the oyster pathogen Haplosporidium nelsoni. J. Eukaryot. Microbiol. 1995, 42, 350–357. [Google Scholar] [CrossRef]
- Stokes, N.; Siddall, M.; Burreson, E. Detection of Haplosporidium nelsoni (Haplosporidia: Haplosporidiidae) in oysters by PCR amplification. Dis. Aquat. Organ. 1995, 23, 145–152. [Google Scholar] [CrossRef]
- Day, J.M.; Franklin, D.E.; Brown, B.L. Use of Competitive PCR to Detect and Quantify Haplosporidium nelsoni Infection (MSX disease) in the Eastern Oyster (Crassostrea virginica). Mar. Biotechnol. 2000, 2, 456–465. [Google Scholar] [CrossRef]
- Carrasco, N.; Andree, K.B.; Lacuesta, B.; Roque, A.; Rodgers, C.; Furones, M.D. Molecular characterization of the Marteilia parasite infecting the common edible cockle Cerastoderma edule in the Spanish Mediterranean coast: A new Marteilia species affecting bivalves in Europe? Aquaculture 2012, 324, 20–26. [Google Scholar] [CrossRef]
- Pernas, M.; Novoa, B.; Berthe, F.; Tafalla, C.; Figueras, A. Molecular methods for the diagnosis of Marteilia refringens. Bull.-Eur. Assoc. Fish Pathol. 2001, 21, 200–208. [Google Scholar]
- Carrasco, N.; Voorbergen-Laarman, M.; Lacuesta, B.; Furones, D.; Engelsma, M.Y. Application of a competitive real time PCR for detection of Marteilia refringens genotype “O” and “M” in two geographical locations: The Ebro Delta, Spain and the Rhine-Meuse Delta, the Netherlands. J. Invertebr. Pathol. 2017, 149, 51–55. [Google Scholar] [CrossRef]
- Berthe, F.; Le Roux, F.; Peyretaillade, E.; Peyret, P.; Rodriguez, D.; Gouy, M.; Vivarès, C.P. The existence of the phylum Paramyxea Desportes and Perkins, 1990 is validated by the phylogenetic analysis of the Marteilia refringens small subunit ribosomal RNA. J. Euk. Microbiol. 2000, 47, 288–293. [Google Scholar] [CrossRef]
- Anderson, T.J.; Adlard, R.D.; Lester, R.J.G. Molecular diagnosis of Marteilia sydneyi (Paramyxea) in Sydney rock oysters, Saccostrea commercialis (Angas). J. Fish. Dis. 1995, 18, 507–510. [Google Scholar] [CrossRef]
- Kleeman, S.N.; LE Roux, F.; Berthe, F.; Adlard, R.D. Specificity of PCR and in situ hybridization assays designed for detection of Marteilia sydneyi and M. refringens. Parasitology 2002, 125, 131–141. [Google Scholar] [CrossRef]
- Yanin, L.; Kang, H.-S.; Hong, H.-K.; Jeung, H.-D.; Kim, B.-K.; Le, T.C.; Kim, Y.-O.; Choi, K.-S. Molecular and histological identification of Marteilioides infection in Suminoe Oyster Crassostrea ariakensis, Manila Clam Ruditapes philippinarum and Pacific Oyster Crassostrea gigas on the south coast of Korea. J. Invertebr. Pathol. 2013, 114, 277–284. [Google Scholar] [CrossRef]
- Itoh, N.; Tun, K.L.; Komiyama, H.; Ueki, N.; Ogawa, K. An ovarian infection in the Iwagaki oyster, Crassostrea nippona, with the protozoan parasite Marteilioides chungmuensis. J. Fish. Dis. 2004, 27, 311–314. [Google Scholar] [CrossRef]
- Carnegie, R.B.; Meyer, G.R.; Blackbourn, J.; Cochennec-Laureau, N.; Berthe, F.C.J.; Bower, S.M. Molecular detection of the oyster parasite Mikrocytos mackini, and a preliminary phylogenetic analysis. Dis. Aquat. Organ. 2003, 54, 219–227. [Google Scholar] [CrossRef]
- Meyer, G.R.; Bower, S.M.; Carnegie, R.B. Sensitivity of a digoxigenin-labelled DNA probe in detecting Mikrocytos mackini, causative agent of Denman Island disease (mikrocytosis), in oysters. J. Invertebr. Pathol. 2005, 88, 89–94. [Google Scholar] [CrossRef]
- Marsh, A.G.; Gauthier, J.D.; Vasta, G.R. A semiquantitative PCR assay for assessing Perkinsus marinus infections in the eastern oyster, Crassostrea virginica. J. Parasitol. 1995, 81, 577–583. [Google Scholar] [CrossRef]
- Ramilo, A.; Pintado, J.; Villalba, A.; Abollo, E. Perkinsus olseni and P. chesapeaki detected in a survey of perkinsosis of various clam species in Galicia (NW Spain) using PCR–DGGE as a screening tool. J. Invertebr. Pathol. 2016, 133, 50–58. [Google Scholar] [CrossRef]
- De la Herrán, R.; Garrido-Ramos, M.A.; Navas, J.I.; Rejón, C.R.; Rejón, M.R. Molecular characterization of the ribosomal RNA gene region of Perkinsus atlanticus: Its use in phylogenetic analysis and as a target for a molecular diagnosis. Parasitology 2000, 120, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.A.; Burreson, E.M.; Reece, K.S. Advanced Perkinsus marinus infections in Crassostrea ariakensis maintained under laboratory conditions. J. Shellfish. Res. 2006, 25, 65–72. [Google Scholar] [CrossRef]
- Abollo, E.; Casas, S.M.; Ceschia, G.; Villalba, A. Differential diagnosis of Perkinsus species by polymerase chain reaction-restriction fragment length polymorphism assay. Mol. Cell. Probes 2006, 20, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Reece, K.S.; Scott, G.P.; Dang, C.; Dungan, C.F. A novel monoclonal Perkinsus chesapeaki in vitro isolate from an Australian cockle, Anadara trapezia. J. Invertebr. Pathol. 2017, 148, 86–93. [Google Scholar] [CrossRef]
- Kim, H.J.; Jun, J.W.; Giri, S.S.; Yun, S.; Kim, S.G.; Kang, J.W.; Han, S.J.; Kwon, J.; Oh, W.T.; Jeon, H.B.; et al. Mass mortality in Korean bay scallop (Argopecten irradians) associated with Ostreid Herpesvirus-1 μVar. Transbound. Emerg. Dis. 2019, 66, 1442–1448. [Google Scholar] [CrossRef]
- Roque, A.; Lopez-Joven, C.; Lacuesta, B.; Elandaloussi, L.; Wagley, S.; Furones, M.D.; Ruiz-Zarzuela, I.; de Blas, I.; Rangdale, R.; Gomez-Gil, B. Detection and identification of tdh-and trh-positive Vibrio parahaemolyticus strains from four species of cultured bivalve molluscs on the Spanish Mediterranean Coast. Appl. Environ. Microbiol. 2009, 75, 7574–7577. [Google Scholar] [CrossRef]
- Schikorski, D.; Renault, T.; Paillard, C.; Bidault-Toffin, A.; Tourbiez, D.; Saulnier, D. Development of TaqMan real-time PCR assays for monitoring Vibrio harveyi infection and a plasmid harbored by virulent strains in European abalone Haliotis tuberculata aquaculture. Aquaculture 2013, 392–395, 106–112. [Google Scholar] [CrossRef]
- Ozanich, R.M.; Colburn, H.A.; Victry, K.D.; Bartholomew, R.A.; Arce, J.S.; Heredia-Langner, A.; Jarman, K.; Kreuzer, H.W.; Bruckner-Lea, C.J. Evaluation of PCR systems for field screening of Bacillus anthracis. Health Secur. 2017, 15, 70–80. [Google Scholar] [CrossRef]
- Stagg, H.M.; Merino, R.; Oladimeji, P.; Steiropoulos, N.; Munro, E.S. Preliminary validation of a portable real-time RT-PCR assay for the detection of salmonid alphavirus. Bull. Eur. Assoc. Fish Pathol. 2018, 38, 121–128. [Google Scholar]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Gao, F.; Jiang, J.-Z.; Wang, J.-Y.; Wei, H.-Y. Real-time isothermal detection of Abalone herpes-like virus and red-spotted grouper nervous necrosis virus using recombinase polymerase amplification. J. Virol. Methods 2018, 251, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, S.; Bai, C.-M.; Yue, Z.; Wang, C.; Yin, W.; Cai, S.; Huang, J. Establishment and application of a cross priming isothermal amplification technique for detection of SB strain of Ostreid herpesvirus-1. J. Fish. China 2015, 39, 580–588. [Google Scholar]
- Pang, Y.; Xie, Z.; Xie, L.; Xie, Z.; Deng, X.; Liu, J.; Peng, Y.; Fan, Q. Establishment and application of visual LAMP assay on Perkinsus olseni. Southwest China J. Agric. Sci. 2012, 25, 302–305. [Google Scholar]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, e63. [Google Scholar] [CrossRef]
- Fakruddin, M. Loop mediated isothermal amplification (LAMP)–an alternative to polymerase chain reaction (PCR). Bangladesh Res. Pub. J. 2011, 5, 425–439. [Google Scholar]
- Tao, Y.; Yun, J.; Wang, J.; Xu, P.; Li, C.; Liu, H.; Lan, Y.; Pan, J.; Du, W. High-performance detection of Mycobacterium bovis in milk using digital LAMP. Food Chem. 2020, 327, 126945. [Google Scholar] [CrossRef]
- Chen, M.; Kuo, S.; Renault, T.; Chang, P. The development of a loop-mediated isothermal amplification assay for rapid and sensitive detection of abalone herpesvirus DNA. J. Virol. Methods 2014, 196, 199–203. [Google Scholar] [CrossRef]
- Feng, C.; Wang, C.; Lin, X.; Zhang, Y.; Lv, J.; Deng, J.; Yuan, X.; Mei, L.; Wu, S. Development of a loop-mediated isothermal amplification method for detection of Perkinsus spp. in mollusks. Dis. Aquat. Organ. 2013, 104, 141–148. [Google Scholar] [CrossRef]
- Wang, D.; Yu, J.; Wang, Y.; Zhang, M.; Li, P.; Liu, M.; Liu, Y. Development of a real-time loop-mediated isothermal amplification (LAMP) assay and visual LAMP assay for detection of African swine fever virus (ASFV). J. Virol. Methods 2020, 276, 113775. [Google Scholar] [CrossRef]
- Biswas, G.; Sakai, M. Loop-mediated isothermal amplification (LAMP) assays for detection and identification of aquaculture pathogens: Current state and perspectives. Appl. Microbiol. Biotechnol. 2014, 98, 2881–2895. [Google Scholar] [CrossRef]
- Zhong, J.; Zhao, X. Isothermal Amplification Technologies for the Detection of Foodborne Pathogens. Food Anal. Methods 2018, 11, 1543–1560. [Google Scholar] [CrossRef]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Recombinase Polymerase Amplification for Diagnostic Applications. Clin. Chem. 2016, 62, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. TrAC Trends Anal. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Yang, X.; Hu, T.; Jiao, B.; Xu, Y.; Zheng, X.; Shen, D. Comparative evaluation of a novel recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay, LAMP, conventional PCR, and leaf-disc baiting methods for detection of Phytophthora sojae. Front. Microbiol. 2019, 10, 1884. [Google Scholar] [CrossRef]
- TwistDx. Recombinase Polymerase Amplification, or RPA, Is the Breakthrough, Isothermal Replacement to PCR. Available online: https://www.twistdx.co.uk/rpa/ (accessed on 29 May 2025).
- Xu, G.; Hu, L.; Zhong, H.; Wang, H.; Yusa, S.-I.; Weiss, T.C.; Romaniuk, P.J.; Pickerill, S.; You, Q.; Zhong, H.; et al. Cross priming amplification: Mechanism and optimization for isothermal DNA amplification. Sci. Rep. 2012, 2, 246. [Google Scholar] [CrossRef]
- Kumar, Y. Isothermal amplification-based methods for assessment of microbiological safety and authenticity of meat and meat products. Food Control 2021, 121, 107679. [Google Scholar] [CrossRef]
- Fang, R.; Li, X.; Hu, L.; You, Q.; Li, J.; Wu, J.; Xu, P.; Zhong, H.; Luo, Y.; Mei, J.; et al. Cross-Priming Amplification for Rapid Detection of Mycobacterium tuberculosis in Sputum Specimens. J. Clin. Microbiol. 2009, 47, 845–847. [Google Scholar] [CrossRef]
- Bai, Z.; Xie, H.; You, Q.; Pickerill, S.; Zhang, Y.; Li, T.; Geng, J.; Hu, L.; Shan, H.; Di, B. Isothermal cross-priming amplification implementation study. Lett. Appl. Microbiol. 2015, 60, 205–209. [Google Scholar] [CrossRef]
- Xu, Z.; Luo, Y.; Soteyome, T.; Lin, C.-W.; Xu, X.; Mao, Y.; Su, J.; Liu, J. Rapid Detection of Food-Borne Escherichia Coli O157:H7 with Visual Inspection by Crossing Priming Amplification (CPA). Food Anal. Methods 2020, 13, 474–481. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Ma, A.; Li, D.; Ye, C. Rapid and sensitive detection of Listeria monocytogenes by cross-priming amplification of lmo0733 gene. FEMS Microbiol. Lett. 2014, 361, 43–51. [Google Scholar] [CrossRef]
- Long, N.; Qiao, Y.; Xu, Z.; Tu, J.; Lu, Z. Recent advances and application in whole-genome multiple displacement amplification. Quant. Biol. 2020, 8, 279–294. [Google Scholar] [CrossRef]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Nalt. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Arneson, N.; Done, S.; Squire, J. The use of whole genome amplification in the study of human disease. Prog. Biophys. Mol. Biol. 2005, 88, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Konakandla, B.; Park, Y.; Margolies, D. Whole genome amplification of Chelex-extracted DNA from a single mite: A method for studying genetics of the predatory mite Phytoseiulus persimilis. Exp. Appl. Acarol. 2006, 40, 241–247. [Google Scholar] [CrossRef]
- Lepere, C.; Demura, M.; Kawachi, M.; Romac, S.; Probert, I.; Vaulot, D. Whole-genome amplification (WGA) of marine photosynthetic eukaryote populations. FEMS Microbiol. Ecol. 2011, 76, 513–523. [Google Scholar] [CrossRef]
- Sabina, J.; Leamon, J.H. Bias in whole genome amplification: Causes and considerations. Whole Genome Amplif. Methods Protoc. 2015, 1347, 15–41. [Google Scholar] [CrossRef]
- Berthet, N.; Reinhardt, A.K.; Leclercq, I.; van Ooyen, S.; Batéjat, C.; Dickinson, P.; Stamboliyska, R.; Old, I.G.; Kong, K.A.; Dacheux, L.; et al. Phi29 polymerase based random amplification of viral RNA as an alternative to random RT-PCR. BMC Mol. Biol. 2008, 9, 77. [Google Scholar] [CrossRef]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu. Rev. Genom. Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef]
- Ren, W.; Wang, C.; Cai, Y. Loop-mediated isothermal amplification for rapid detection of acute viral necrobiotic virus in scallop Chlamys farreri. Acta Virol. 2009, 53, 161–167. [Google Scholar] [CrossRef]
- Jiang, J.; Yang, R.; Zhang, H.; Liu, G.; Zhu, Z.; Su, Y.; Wu, E.; Wang, J. Detection of abalone shrivelling syndrome-associated virus using loop-mediated isothermal amplification. J. Fish Dis. 2013, 37, 63–67. [Google Scholar] [CrossRef]
- Ren, W.; Renault, T.; Cai, Y.; Wang, C. Development of a loop-mediated isothermal amplification assay for rapid and sensitive detection of ostreid herpesvirus 1 DNA. J. Virol. Methods 2010, 170, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zaczek-Moczydłowska, M.A.; Mohamed-Smith, L.; Toldrà, A.; Hooper, C.; Campàs, M.; Furones, M.D.; Bean, T.P.; Campbell, K. A single-tube HNB-based loop-mediated isothermal amplification for the robust detection of the Ostreid herpesvirus 1. Int. J. Mol. Sci. 2020, 21, 6605. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Jiang, J.-Z.; Wang, J.-Y.; Wei, H.-Y. Real-time quantitative isothermal detection of Ostreid herpesvirus-1 DNA in Scapharca subcrenata using recombinase polymerase amplification. J. Virol. Methods 2018, 255, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Toldrà, A.; Furones, M.D.; O’Sullivan, C.K.; Campàs, M. Detection of isothermally amplified ostreid herpesvirus 1 DNA in Pacific oyster (Crassostrea gigas) using a miniaturised electrochemical biosensor. Talanta 2020, 207, 120308. [Google Scholar] [CrossRef]
- Xia, J.; Bai, C.; Wang, C.; Song, X.; Huang, J. Complete genome sequence of Ostreid herpesvirus-1 associated with mortalities of Scapharca broughtonii broodstocks. Virol. J. 2015, 12, 110. [Google Scholar] [CrossRef]
- Cano, I.; Wood, G.; Stone, D.; Noyer, M.; Canier, L.; Arzul, I. Loop-Mediated Isothermal Amplification for the Fast Detection of Bonamia ostreae and Bonamia exitiosa in Flat Oysters. Pathogens 2024, 13, 132. [Google Scholar] [CrossRef]
- Xie, L.; Xie, Z.; Pang, Y.; Deng, X.; Xie, Z.; Liu, J. Development of a loop-mediated isothermal amplification assay for visual detection of Marteilia refringens in shellfish. Chin. J. Vet. Sci. 2012, 32, 993–996. [Google Scholar]
- Mendoza-Avilés, I.; Muñoz-Rojas, C.A.; Rojas, M.; Estrada, N. Loop-mediated isothermal amplification for diagnosing marine pathogens in tissues of Crassostrea spp. and white shrimp, Litopenaeus vannamei, farmed in Mexico. Cienc. Mar. 2021, 47, 227–239. [Google Scholar] [CrossRef]
- Wu, L.; Ye, L.; Wang, Z.; Cui, Y.; Wang, J. Utilization of recombinase polymerase amplification combined with a lateral flow strip for detection of Perkinsus beihaiensis in the oyster Crassostrea hongkongensis. Parasit. Vectors. 2019, 12, 360. [Google Scholar] [CrossRef]
- Qu, P.; Wang, C.-M.; Ren, W.-C.; Liang, Y.-T.; Jia, Z.-L.; Huang, J.; Pan, L.-Q. Establishment and application of a loop-mediated isothermal amplification (LAMP) method for Perkinsus olseni detection. J. Fish. China 2012, 36, 1281–1289. [Google Scholar] [CrossRef]
- Bass, D.; Stentiford, G.D.; Littlewood, D.; Hartikainen, H. Diverse Applications of Environmental DNA Methods in Parasitology. Trends Parasitol. 2015, 31, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.; Wight, N.; Crosson, L.; White, S.; Strenge, R. Validation of a quantitative PCR assay for detection and quantification of ‘Candidatus Xenohaliotis californiensis’. Dis. Aquat. Organ. 2014, 108, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Vincent-Hubert, F.; Morga, B.; Renault, T.; Le Guyader, F. Adsorption of norovirus and ostreid herpesvirus type 1 to polymer membranes for the development of passive samplers. J. Appl. Microbiol. 2017, 122, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Toldrà, A.; Toldrà, A.; Andree, K.B.; Andree, K.B.; Bertomeu, E.; Bertomeu, E.; Roque, A.; Roque, A.; Carrasco, N.; Carrasco, N.; et al. Rapid capture and detection of ostreid herpesvirus-1 from Pacific oyster Crassostrea gigas and seawater using magnetic beads. PLoS ONE 2018, 13, e0205207. [Google Scholar] [CrossRef]
- Morga, B.; Faury, N.; Guesdon, S.; Chollet, B.; Renault, T. Haemocytes from Crassostrea gigas and OsHV-1: A promising in vitro system to study host/virus interactions. J. Invertebr. Pathol. 2017, 150, 45–53. [Google Scholar] [CrossRef]
- Sakudo, A.; Onodera, T. Virus capture using anionic polymer-coated magnetic beads. Int. J. Mol. Med. 2012, 30, 3–7. [Google Scholar] [CrossRef]
- Sakudo, A.; Masrinoul, P.; Tanaka, Y.; Ikuta, K. Capture of dengue virus type 3 using anionic polymer-coated magnetic beads. Int. J. Mol. Med. 2011, 28, 625–628. [Google Scholar] [CrossRef]
- Chomczynski, P.; Rymaszewski, M. Alkaline polyethylene glycol-based method for direct PCR from bacteria, eukaryotic tissue samples, and whole blood. Biotechniques 2006, 40, 454–458. [Google Scholar] [CrossRef]
- Dimitrakopoulou, M.-E.; Stavrou, V.; Kotsalou, C.; Vantarakis, A. Boiling extraction method vs commercial kits for bacterial DNA isolation from food samples. J. Food Nutr. Res. 2020, 3, 311–319. [Google Scholar] [CrossRef]
- Barreda-García, S.; Miranda-Castro, R.; De-Los-Santos-Álvarez, N.; Miranda-Ordieres, A.J.; Lobo-Castañón, M.J. Helicase-dependent isothermal amplification: A novel tool in the development of molecular-based analytical systems for rapid pathogen detection. Anal. Bioanal. Chem. 2018, 410, 679–693. [Google Scholar] [CrossRef]
- Nagamine, K.; Hase, T.; Notomi, T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol. Cell. Probes 2002, 16, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Zhang, H.; Liang, H.; Gong, G.; Cai, Y. Betaine-assisted recombinase polymerase assay for rapid hepatitis B virus detection. Biotechnol. Appl. Biochem. 2021, 68, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Pasookhush, P.; Longyant, S.; Sithigorngul, P.; Chaivisuthangkura, P. Development of duplex loop-mediated isothermal amplification (dLAMP) combined with lateral flow dipstick (LFD) for the rapid and specific detection of Vibrio vulnificus and V. parahaemolyticus. N. Am. J. Aquac. 2016, 78, 327–336. [Google Scholar] [CrossRef]
- Surasilp, T.; Longyant, S.; Rukpratanporn, S.; Sridulyakul, P.; Sithigorngul, P.; Chaivisuthangkura, P. Rapid and sensitive detection of Vibrio vulnificus by loop-mediated isothermal amplification combined with lateral flow dipstick targeted to rpoS gene. Mol. Cell. Probes 2011, 25, 158–163. [Google Scholar] [CrossRef]
- Chu, F.-L.; Hale, R. Relationship between pollution and susceptibility to infectious disease in the eastern oyster, Crassostrea virginica. Mar. Environ. Res. 1994, 38, 243–256. [Google Scholar] [CrossRef]
- Renault, T. Viruses infecting marine mollusks. In Studies in Viral Ecol; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2021; pp. 275–303. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, H.; Zerna, G.; Knox, A.; Ackerly, D.; Agius, J.; Helbig, K.; Beddoe, T. Enhancing Biosecurity in Mollusc Aquaculture: A Review of Current Isothermal Nucleic Acid Detection Methods. Animals 2025, 15, 1664. https://doi.org/10.3390/ani15111664
Abbas H, Zerna G, Knox A, Ackerly D, Agius J, Helbig K, Beddoe T. Enhancing Biosecurity in Mollusc Aquaculture: A Review of Current Isothermal Nucleic Acid Detection Methods. Animals. 2025; 15(11):1664. https://doi.org/10.3390/ani15111664
Chicago/Turabian StyleAbbas, Hoda, Gemma Zerna, Alexandra Knox, Danielle Ackerly, Jacinta Agius, Karla Helbig, and Travis Beddoe. 2025. "Enhancing Biosecurity in Mollusc Aquaculture: A Review of Current Isothermal Nucleic Acid Detection Methods" Animals 15, no. 11: 1664. https://doi.org/10.3390/ani15111664
APA StyleAbbas, H., Zerna, G., Knox, A., Ackerly, D., Agius, J., Helbig, K., & Beddoe, T. (2025). Enhancing Biosecurity in Mollusc Aquaculture: A Review of Current Isothermal Nucleic Acid Detection Methods. Animals, 15(11), 1664. https://doi.org/10.3390/ani15111664