

Initial Screening of Extrachromosomal Circular DNA Candidates for Pork Meat Quality Traits Using Circle-Seq and RNA-Seq Analysis
 , , , , and
, , , , and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Description
2.2. Determination of Meat Quality Traits
2.3. Circle-Seq Data Analysis
2.4. EccDNAs Difference Analysis and Gene Annotation
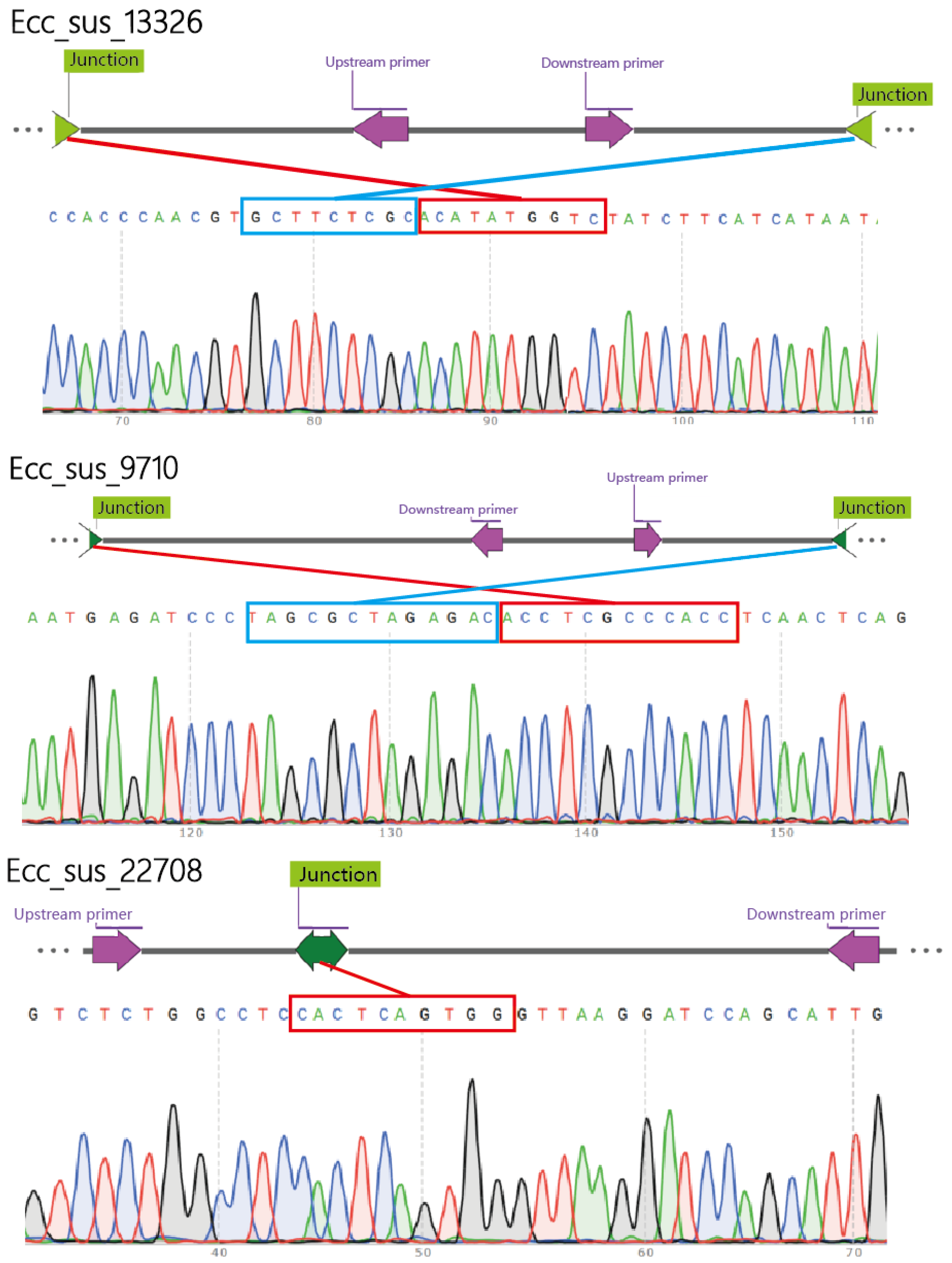
2.5. Verification of eccDNAs
2.6. RNA-Seq Data Analysis
2.7. Validation of DEGs
2.8. Enrichment Analysis and Functional Annotation of eccDNAs and eccDEGs
2.9. Prediction of Regulatory Network of eccDNAs-miRNAs-Genes
3. Results
3.1. Comparison of Meat Quality Indicators in the YN and YL Groups
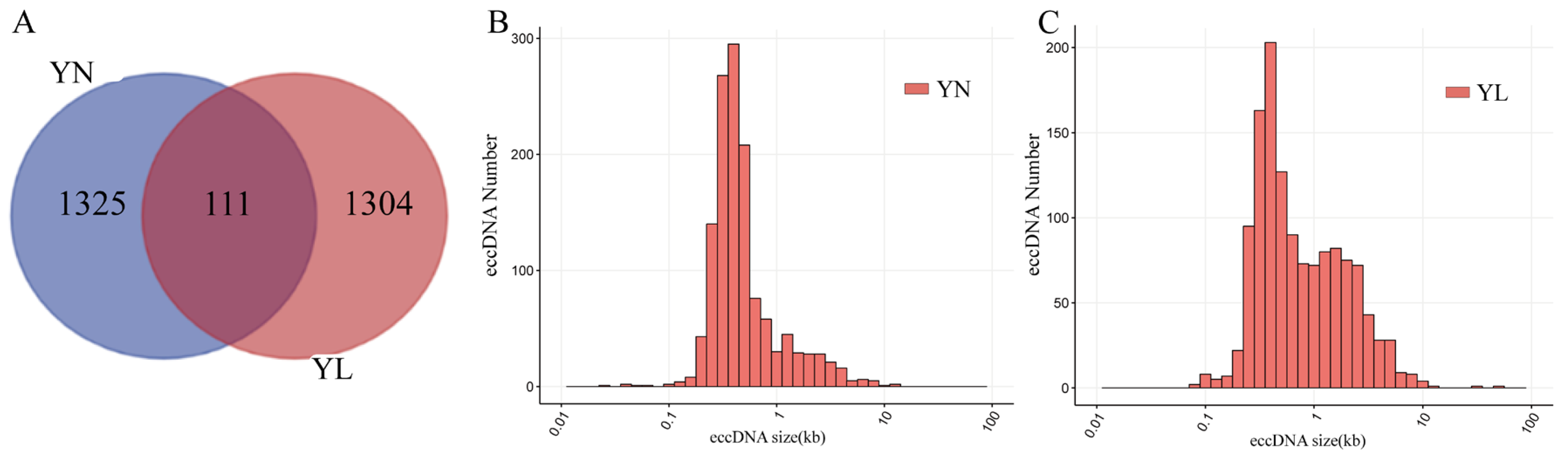
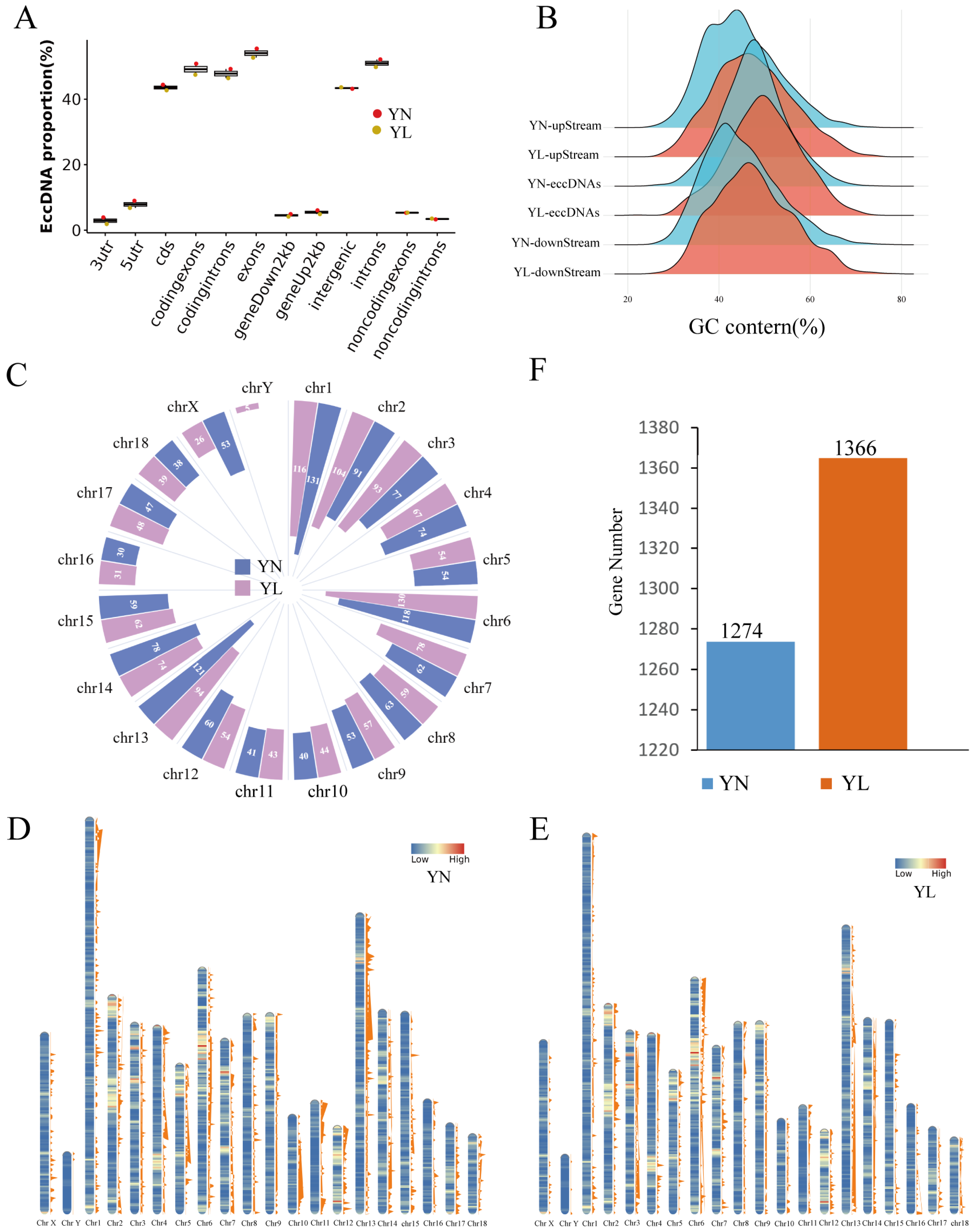
3.2. Basic Features of eccDNAs in YN and YL Pigs
3.3. Validation of eccDNAs
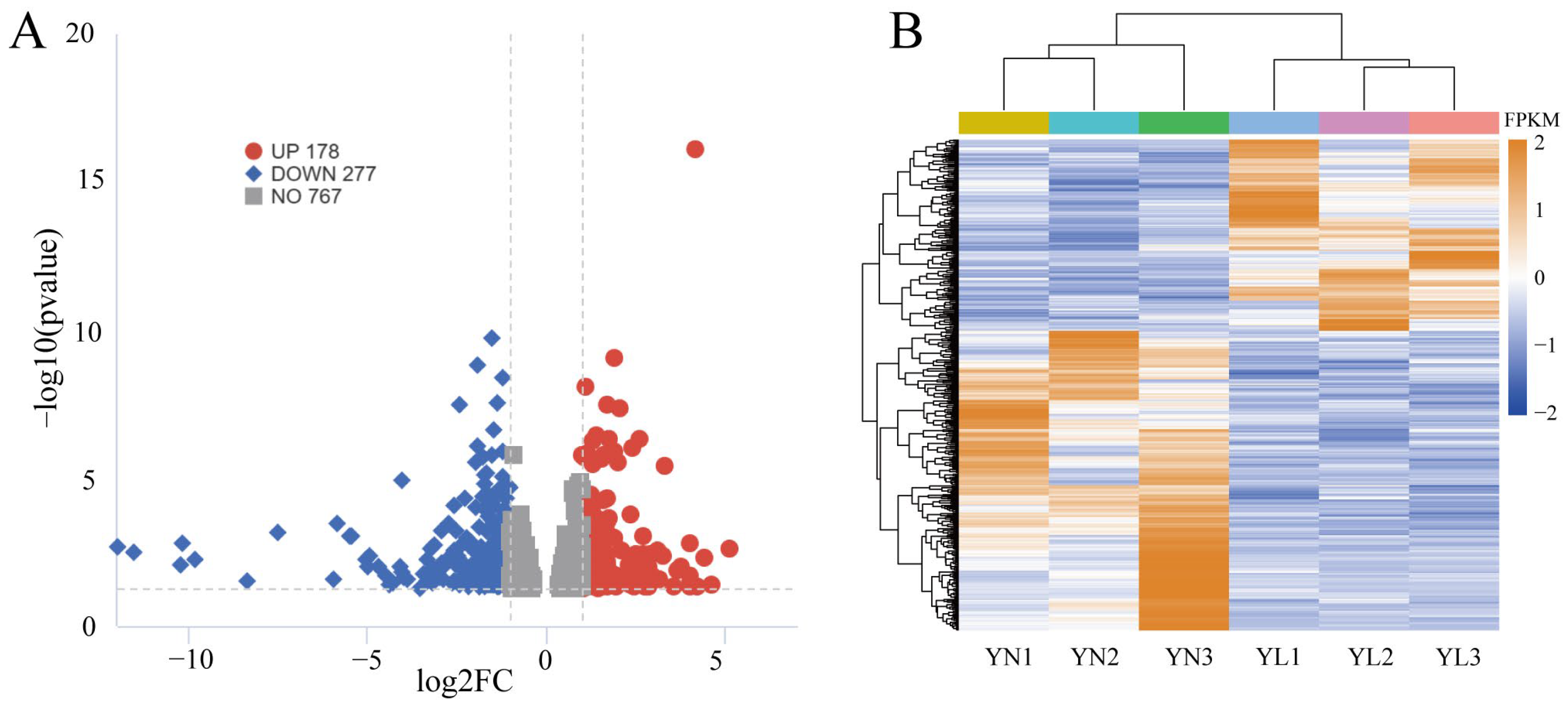
3.4. Identification of DEGs
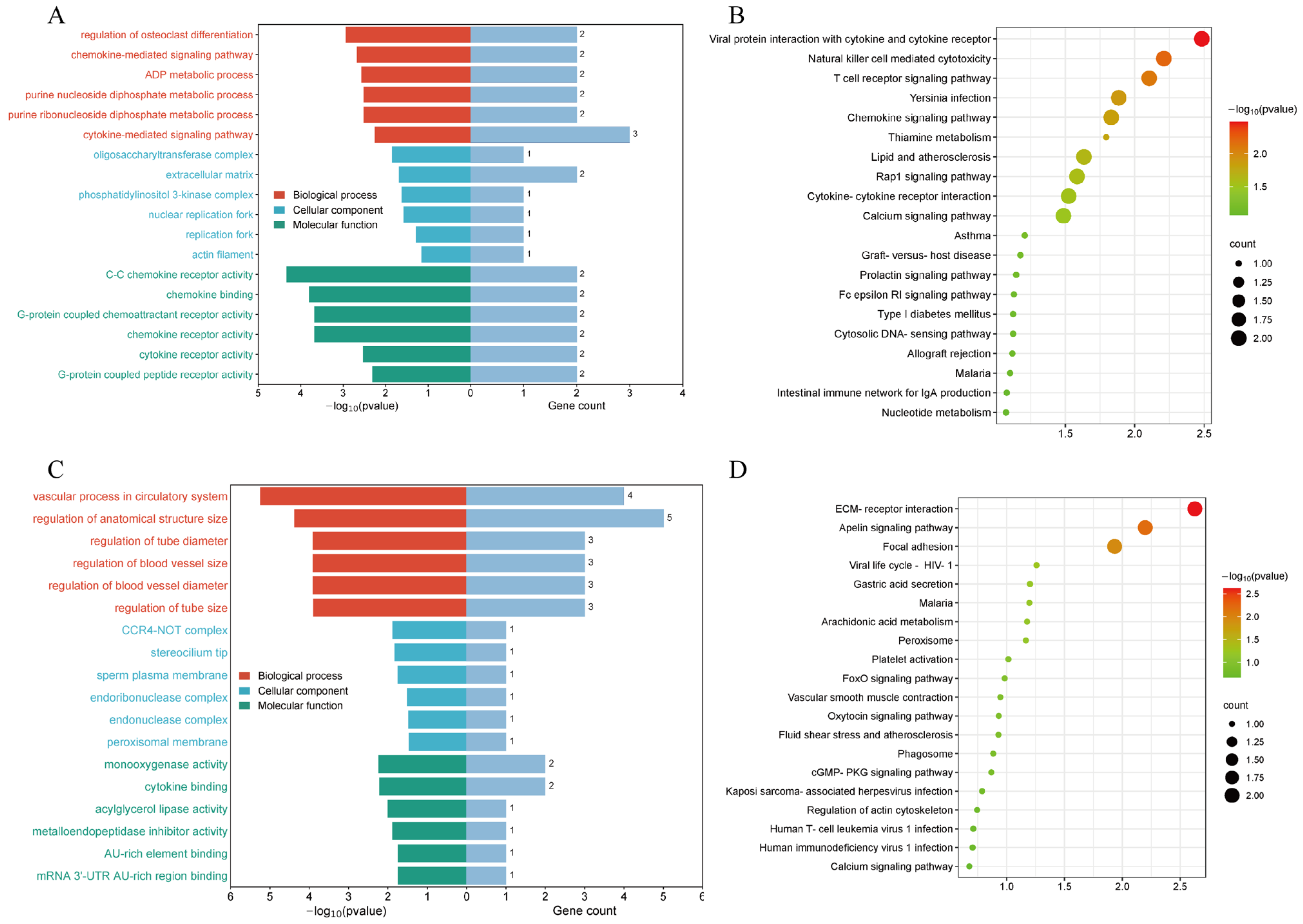
3.5. Identification and Enrichment Analysis of Differentially Expressed eccDNA-Amplified Encoding Genes
3.6. EccDNAs–miRNAs–Genes Regulatory Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, Y.; Chen, S.; Yuan, J.; Shi, Y.; Wang, Y.; Xi, Y.; Qi, X.; Guo, Y.; Sheng, X.; Liu, J.; et al. Comprehensive Analysis of the lncRNA-miRNA-mRNA Regulatory Network for Intramuscular Fat in Pigs. Genes 2023, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Li, L.; Yu, N.; Xiong, S.; Xiao, S.; Zheng, H.; Huang, Z.; Guo, Y.; Huang, L. Phenotypic correlations of carpal gland diverticular number with production traits and its genome-wide association analysis in multiple pig populations. Anim. Genet. 2024, 55, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xie, F.; Wang, J.; Luo, J.; Chen, T.; Jiang, Q.; Xi, Q.; Liu, G.E.; Zhang, Y. Integrated meta-omics reveals the regulatory landscape involved in lipid metabolism between pig breeds. Microbiome 2024, 12, 33. [Google Scholar] [CrossRef]
- Ma, C.; Wang, W.; Wang, Y.; Sun, Y.; Kang, L.; Zhang, Q.; Jiang, Y. TMT-labeled quantitative proteomic analyses on the longissimus dorsi to identify the proteins underlying intramuscular fat content in pigs. J. Proteom. 2020, 213, 103630. [Google Scholar] [CrossRef]
- Gerovska, D.; Araúzo-Bravo, M.J. Skeletal Muscles of Sedentary and Physically Active Aged People Have Distinctive Genic Extrachromosomal Circular DNA Profiles. Int. J. Mol. Sci. 2023, 24, 2736. [Google Scholar] [CrossRef]
- Hotta, Y.; Bassel, A. Molecular size and circularity of dna in cells of mammals and higher plants. Proc. Natl. Acad. Sci. USA 1965, 53, 356–362. [Google Scholar] [CrossRef]
- Møller, H.D.; Lin, L.; Xiang, X.; Petersen, T.S.; Huang, J.; Yang, L.; Kjeldsen, E.; Jensen, U.B.; Zhang, X.; Liu, X.; et al. CRISPR-C: Circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res. 2018, 46, e131. [Google Scholar] [CrossRef]
- Wen, A.; Zhang, W.; Xu, J.; Wang, K.; Hu, H. Identification and characterization of extrachromosomal circular DNA in Wei and Large White pigs by high-throughput sequencing. Front. Vet. Sci. 2023, 10, 1085474. [Google Scholar] [CrossRef]
- Xue, Y.; Li, C.; Duan, D.; Wang, M.; Han, X.; Wang, K.; Qiao, R.; Li, X.J.; Li, X.L. Genome-wide association studies for growth-related traits in a crossbreed pig population. Anim. Genet. 2021, 52, 217–222. [Google Scholar] [CrossRef]
- NY/T 821-2019; Technical Code of Practice for Pork Quality Assessment. Ministry of Agriculture and Rural Affairs of the People’s Republic of China: Beijing, China, 2019.
- GB 5009.3-2016; National Food Safety Standards Determination of Moisture in Food. National Health and Family Planning Commission of the People’s Republic of China: Beijing, China, 2017.
- GB 5009.5-2016; National Food Safety Standards Determination of Protein in Food. National Health and Family Planning Commission of the People’s Republic of China State Food and Drug Administration: Beijing, China, 2017.
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:13033997. [Google Scholar]
- Prada-Luengo, I.; Krogh, A.; Maretty, L.; Regenberg, B. Sensitive detection of circular DNAs at single-nucleotide resolution using guided realignment of partially aligned reads. BMC Bioinform. 2019, 20, 663. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG graphics to visualize and map genome-wide data on the idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef]
- Parkhomchuk, D.; Borodina, T.; Amstislavskiy, V.; Banaru, M.; Hallen, L.; Krobitsch, S.; Lehrach, H.; Soldatov, A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009, 37, e123. [Google Scholar] [CrossRef]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Rosenvold, K.; Andersen, H.J. Factors of significance for pork quality-a review. Meat Sci. 2003, 64, 219–237. [Google Scholar] [CrossRef]
- Kumar, S.T.; Zheng, Y.; Xu, J.; Zhao, Z.; Zhang, Q.; Zhang, Y.; Li, M.; Zou, H.; Azeem, R.M.; Sun, W.S.; et al. Transcriptome and Metabolome Insights into Key Genes Regulating Fat Deposition and Meat Quality in Pig Breeds. Animals 2024, 14, 3560. [Google Scholar] [CrossRef]
- Li, F.; Yang, L.; Han, J.; Han, X.; Peng, L.; Du, Y.; Xia, H.; Yang, L.; Zhou, Y. Characterization of extrachromosomal circular DNA in cattle using 676 whole genome sequencing datasets. Anim. Genet. 2022, 53, 761–768. [Google Scholar] [CrossRef]
- Moller, H.D.; Ramos-Madrigal, J.; Prada-Luengo, I.; Gilbert, M.T.P.; Regenberg, B. Near-Random Distribution of Chromosome-Derived Circular DNA in the Condensed Genome of Pigeons and the Larger, More Repeat-Rich Human Genome. Genome Biol. Evol. 2020, 12, 3762–3777. [Google Scholar] [CrossRef]
- Ouyang, Y.; Lu, W.; Wang, Y.; Wang, B.; Li, F.; Li, X.; Bai, Y.; Wang, Y. Integrated analysis of mRNA and extrachromosomal circular DNA profiles to identify the potential mRNA biomarkers in breast cancer. Gene 2023, 857, 147174. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Pan, X.; Lin, L.; Li, L.; Yuan, S.; Han, P.; Ji, X.; Li, H.; Wang, C.; Chu, Z.; et al. Characterization of Plasma Extrachromosomal Circular DNA in Gouty Arthritis. Front. Genet. 2022, 13, 859513. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ma, S.; Zhao, Y.; Guo, D.; Wang, H.; Kuang, M.; Li, X. Identification and characterization of extrachromosomal circular DNA in alcohol induced osteonecrosis of femoral head. Front. Genet. 2022, 13, 918379. [Google Scholar] [CrossRef]
- Ren, S.; Wu, D.; Shen, X.; Wu, Q.; Li, C.; Xiong, H.; Xiong, Z.; Gong, R.; Liu, Z.; Wang, W.; et al. Deciphering the role of extrachromosomal circular DNA in adipose stem cells from old and young donors. Stem. Cell Res. Ther. 2023, 14, 341. [Google Scholar] [CrossRef]
- Durkin, K.; Coppieters, W.; Drögemüller, C.; Ahariz, N.; Cambisano, N.; Druet, T.; Fasquelle, C.; Haile, A.; Horin, P.; Huang, L.; et al. Serial translocation by means of circular intermediates underlies colour sidedness in cattle. Nature 2012, 482, 81–84. [Google Scholar] [CrossRef]
- Guo, H.; Cui, B.D.; Gong, M.; Li, Q.X.; Zhang, L.X.; Chen, J.L.; Chi, J.; Zhu, L.L.; Xu, E.P.; Wang, Z.M.; et al. An ethanolic extract of Arctium lappa L. leaves ameliorates experimental atherosclerosis by modulating lipid metabolism and inflammatory responses through PI3K/Akt and NF-κB singnaling pathways. J. Ethnopharmacol. 2024, 325, 117768. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Quan, C.; Huang, Y.; Ji, W.; Yu, L.; Li, X.; Zhang, Y.; Zheng, Z.; Zou, H.; Li, Q.; et al. Constitutive ERK1/2 activation contributes to production of double minute chromosomes in tumour cells. J. Pathol. 2015, 235, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Dong, Y.; Zhang, C.; Su, B.; Ning, N.; Zhou, S.; Zhang, M.; Huang, Q.; Li, Y.; Wang, S. Zishen Yutai pills restore fertility in premature ovarian failure through regulating arachidonic acid metabolism and the ATK pathway. J. Ethnopharmacol. 2024, 324, 117782. [Google Scholar] [CrossRef]
- Zhu, Y.; Gujar, A.D.; Wong, C.H.; Tjong, H.; Ngan, C.Y.; Gong, L.; Chen, Y.A.; Kim, H.; Liu, J.; Li, M.; et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 2021, 39, 694–707.e697. [Google Scholar] [CrossRef]
- Yao, Y.; Hu, C.; Song, Q.; Li, Y.; Da, X.; Yu, Y.; Li, H.; Clark, I.M.; Chen, Q.; Wang, Q.K. ADAMTS16 activates latent TGF-β, accentuating fibrosis and dysfunction of the pressure-overloaded heart. Cardiovasc. Res. 2020, 116, 956–969. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, H.; Jin, W.; Lee, H.J. Acute exercise regulates adipogenic gene expression in white adipose tissue. Biol. Sport 2016, 33, 381–391. [Google Scholar] [CrossRef]
- Di Cesare, P.E.; Chen, F.S.; Moergelin, M.; Carlson, C.S.; Leslie, M.P.; Perris, R.; Fang, C. Matrix-matrix interaction of cartilage oligomeric matrix protein and fibronectin. Matrix Biol. 2002, 21, 461–470. [Google Scholar] [CrossRef]
- Zhu, M.; Tong, X.; Qiu, Q.; Pan, J.; Wei, S.; Ding, Y.; Feng, Y.; Hu, X.; Gong, C. Identification and characterization of extrachromosomal circular DNA in the silk gland of Bombyx mori. Insect Sci. 2023, 30, 1565–1578. [Google Scholar] [CrossRef]
- Zhu, M.; Pan, J.; Tong, X.; Qiu, Q.; Zhang, X.; Zhang, Y.; Sun, S.; Feng, Y.; Xue, R.; Cao, G.; et al. BmCPV-Derived Circular DNA vcDNA-S7 Mediated by Bombyx mori Reverse Transcriptase (RT) Regulates BmCPV Infection. Front. Immunol. 2022, 13, 861007. [Google Scholar] [CrossRef]
- Jiang, X.; Pan, X.; Li, W.; Han, P.; Yu, J.; Li, J.; Zhang, H.; Lv, W.; Zhang, Y.; He, Y.; et al. Genome-wide characterization of extrachromosomal circular DNA in gastric cancer and its potential role in carcinogenesis and cancer progression. Cell. Mol. Life Sci. 2023, 80, 191. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| eccDNAs | Forward Primers | Reverse Primers |
|---|---|---|
| Ecc_sus_13326 | CAATCGCACCAGTGAGGCC | GGTGGTGTTGAGGATGAATGTG |
| Ecc_sus_9710 | ATTCCCATTGTGGCTCTTG | CACAAATCCAAAGGGAACAA |
| Ecc_sus_22708 | GAGATGTGCCAGGTGGACTGT | GTAATCAACCCTGACCATCTCCT |
| Gene Name | Forward Primers | Reverse Primers |
|---|---|---|
| UBA7 | CTTCTGCTGAGTTTGGCCCT | ATAGTTCTGAGCTCGCAGGC |
| GPCPD1 | TCTGGTGGGAGCTTTGCTTT | TGGTGGGGATTTGTCTTCCAA |
| AKR1C4 | GACATCGAGGTGCAGGGAAT | CAAAGGCTGCACCGTGACTA |
| GRM4 | GTCGGCAGACAGATGGTCTT | TGCTCTTAGGGACCAAATCCC |
| FRAS1 | GTCAAGAAGTGCACCAACCG | CGGCATCGATCACAAACTGC |
| PER2 | ACTTCGTCTTCCTGTCCAGATG | CTTTCAGCTCCCTCAGCGTT |
| Item | YN | YL |
|---|---|---|
| Meat color 45 min | 4.00 ± 0.00 a | 2.40 ± 0.17 b |
| Ph 45 min | 5.45 ± 0.12 b | 6.17 ± 0.15 c |
| Marble grain 45 min | 3.50 ± 0.50 | 2.50 ± 0.50 |
| Drip loss % | 1.04 ± 0.12 | 1.10 ± 0.27 |
| Moisture % | 73.66 ± 1.66 | 75.14 ± 0.66 |
| Intramuscular fat content (dry sample) % | 13.80 ± 0.63 a | 9.39 ± 0.44 c |
| Protein content (dry sample) % | 77.90 ± 0.87 | 81.57 ± 2.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, L.; Wu, J.; Dou, T.; Chu, D.; Li, X.; Han, X.; Qiao, R.; Wang, K.; Yang, F.; Li, X. Initial Screening of Extrachromosomal Circular DNA Candidates for Pork Meat Quality Traits Using Circle-Seq and RNA-Seq Analysis. Animals 2025, 15, 1590. https://doi.org/10.3390/ani15111590
Bai L, Wu J, Dou T, Chu D, Li X, Han X, Qiao R, Wang K, Yang F, Li X. Initial Screening of Extrachromosomal Circular DNA Candidates for Pork Meat Quality Traits Using Circle-Seq and RNA-Seq Analysis. Animals. 2025; 15(11):1590. https://doi.org/10.3390/ani15111590
Chicago/Turabian StyleBai, Liyao, Jiahao Wu, Tengfei Dou, Donghui Chu, Xinjian Li, Xuelei Han, Ruimin Qiao, Kejun Wang, Feng Yang, and Xiuling Li. 2025. "Initial Screening of Extrachromosomal Circular DNA Candidates for Pork Meat Quality Traits Using Circle-Seq and RNA-Seq Analysis" Animals 15, no. 11: 1590. https://doi.org/10.3390/ani15111590
APA StyleBai, L., Wu, J., Dou, T., Chu, D., Li, X., Han, X., Qiao, R., Wang, K., Yang, F., & Li, X. (2025). Initial Screening of Extrachromosomal Circular DNA Candidates for Pork Meat Quality Traits Using Circle-Seq and RNA-Seq Analysis. Animals, 15(11), 1590. https://doi.org/10.3390/ani15111590