
Integrated Analysis of Differential Expression Profiles of miRNA and mRNA in Gonads of Scatophagus argus Provides New Insights into Sexually Biased Gene Expression


 ,
, 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Total RNA Extraction
2.3. Library Preparation and Small RNA (sRNA) Sequencing
2.4. miRNAs Identification
2.5. MiRNA Expression and Differential Expression Analysis
2.6. Verification of miRNA Expression by Real-Time Quantitative PCR (RT-qPCR)
2.7. Target Gene Prediction
2.8. GO and KEGG Enrichment Analysis
3. Results
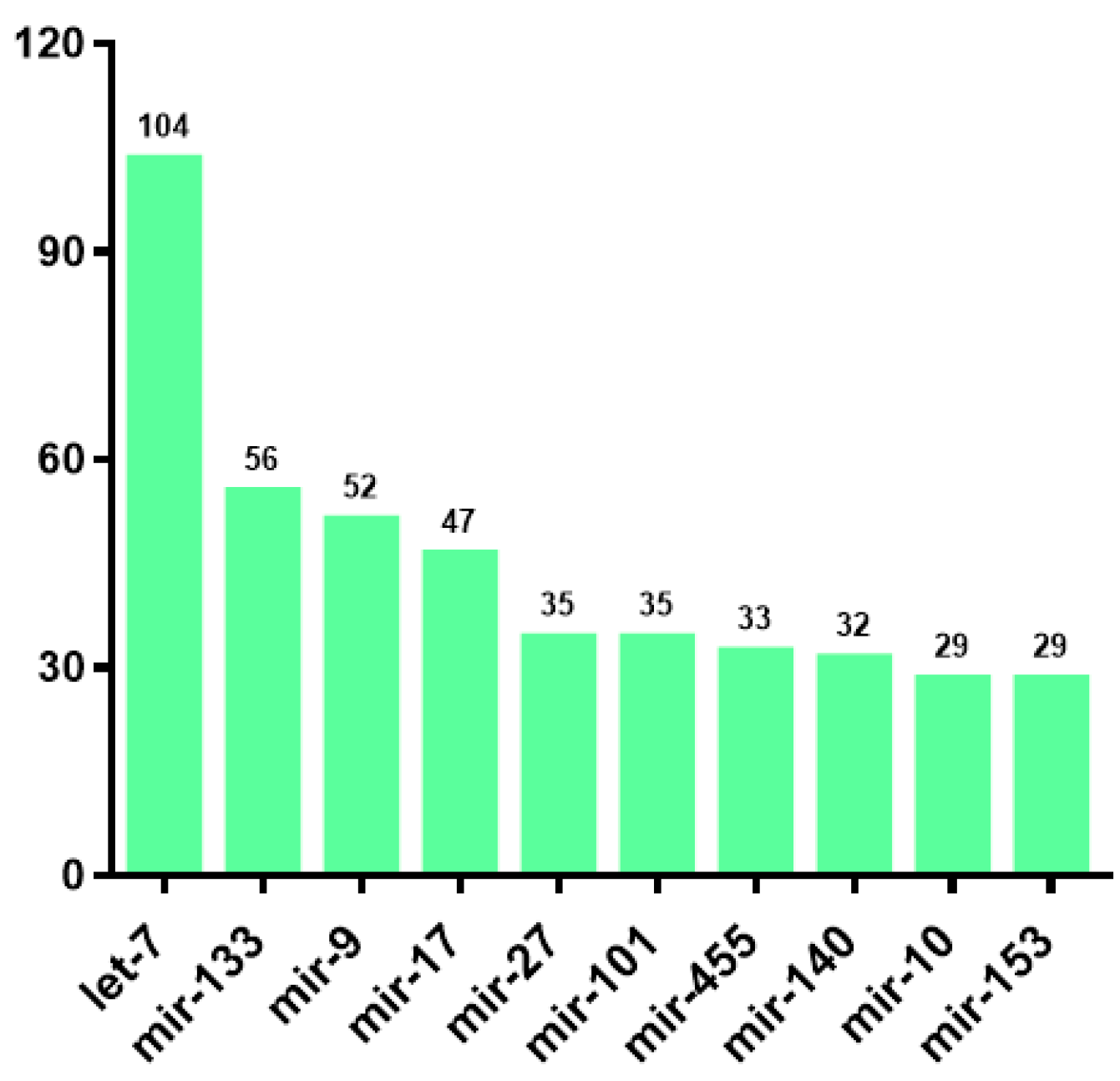
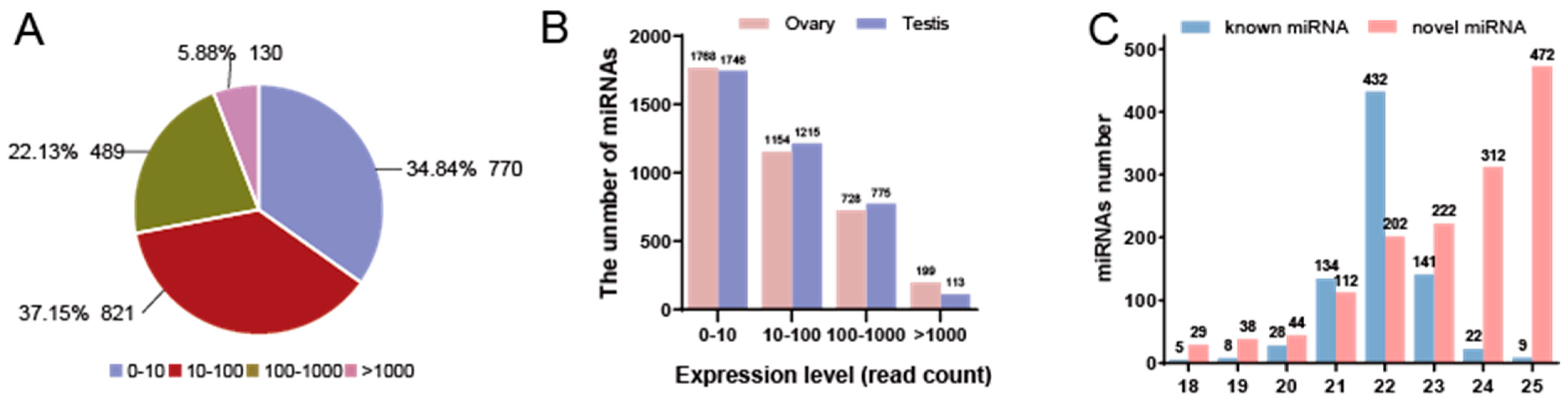
3.1. Construction of miRNA Library
3.2. Identification and Classification of miRNAs
3.3. Identification and Validation of Differentially Expressed miRNAs (DEMs)
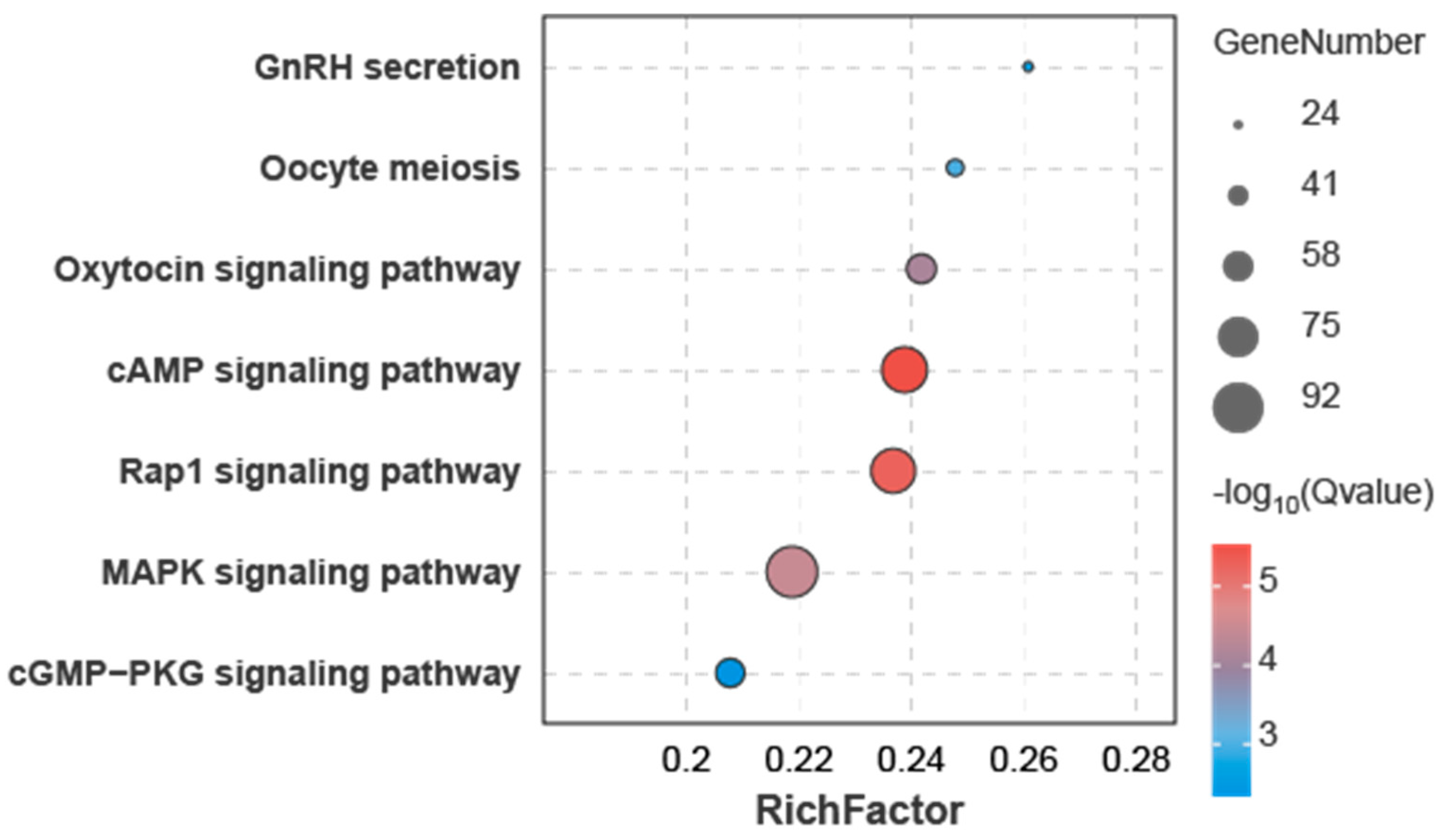
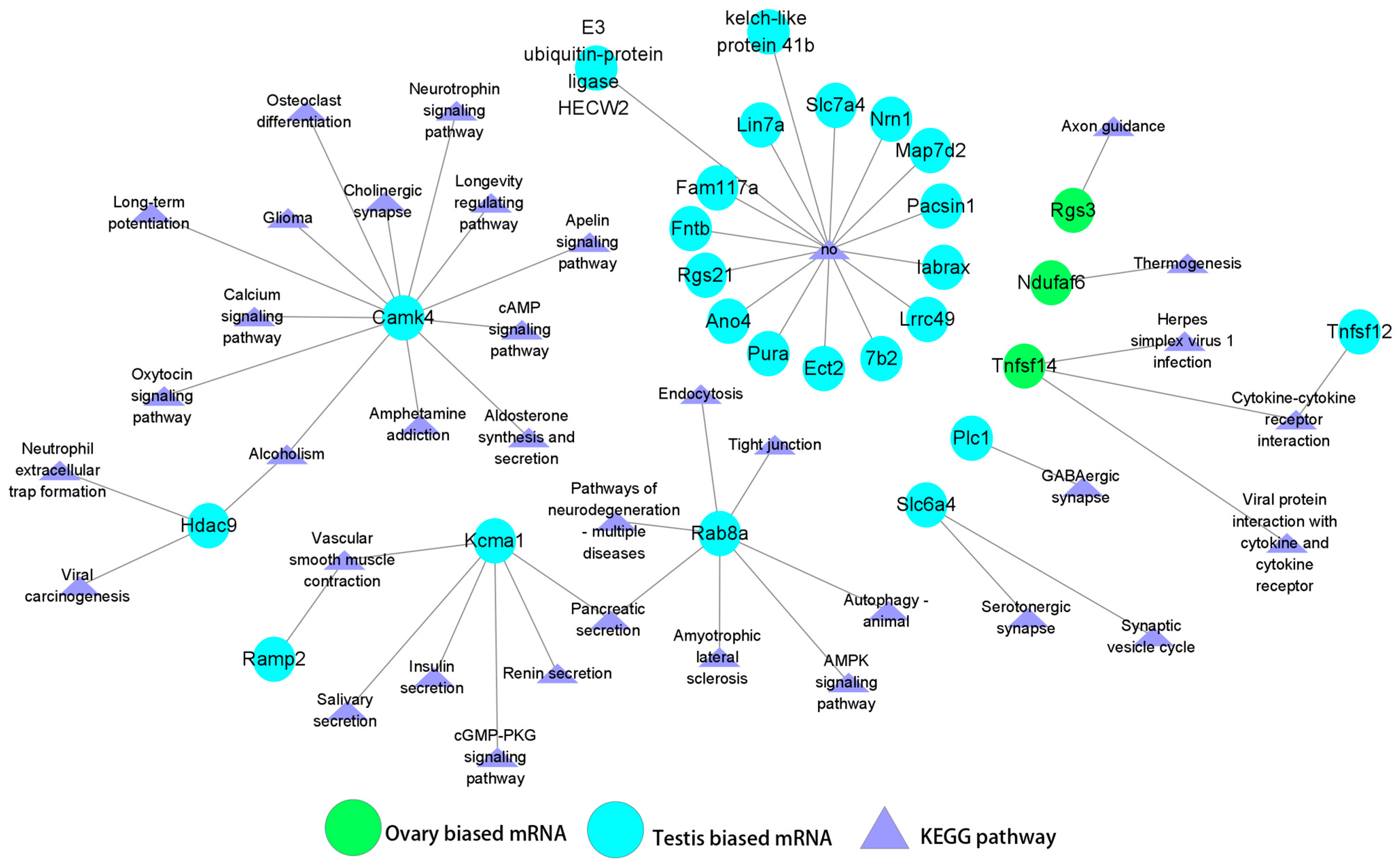
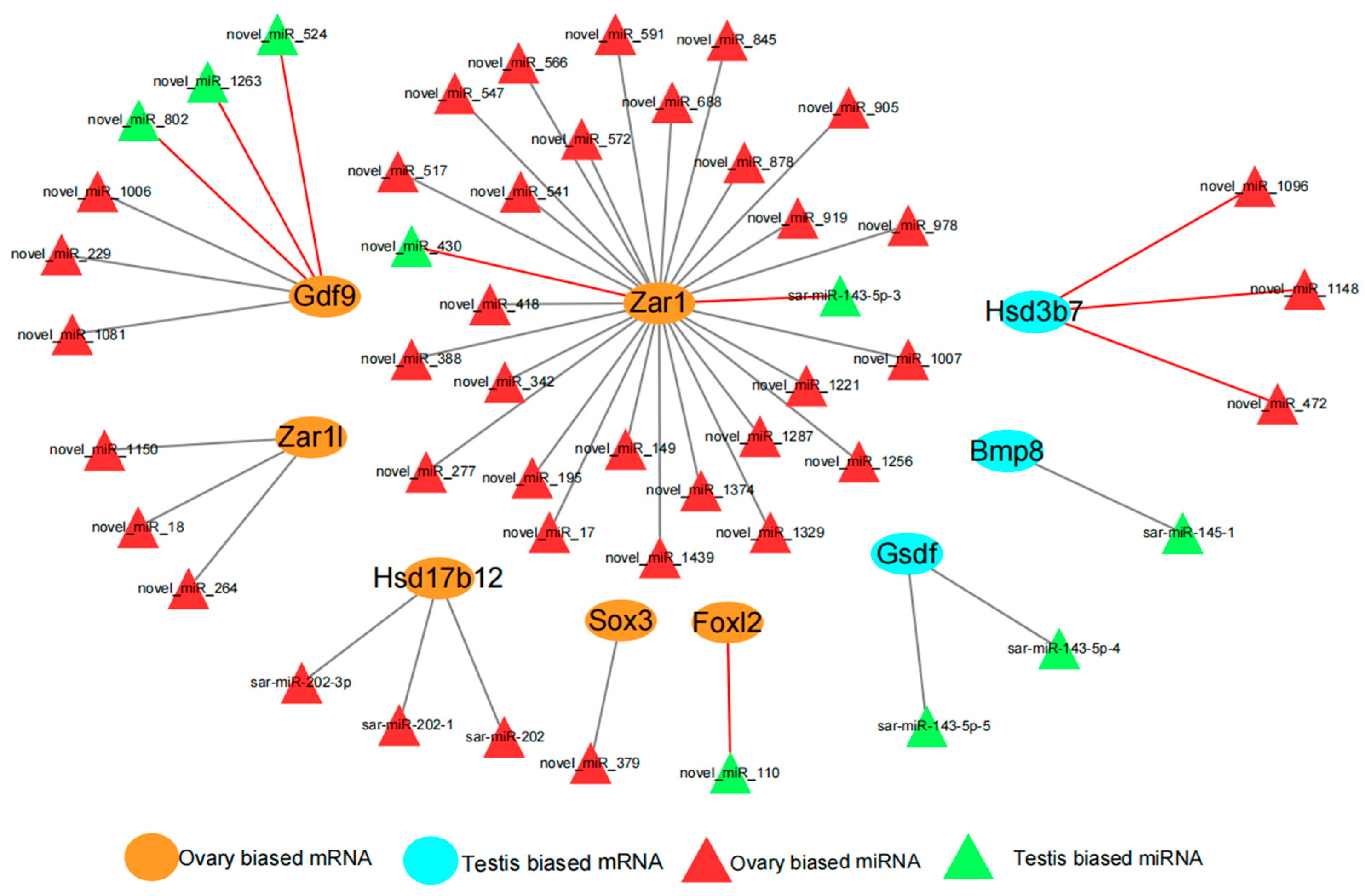
3.4. Joint miRNA and Transcriptome Analysis
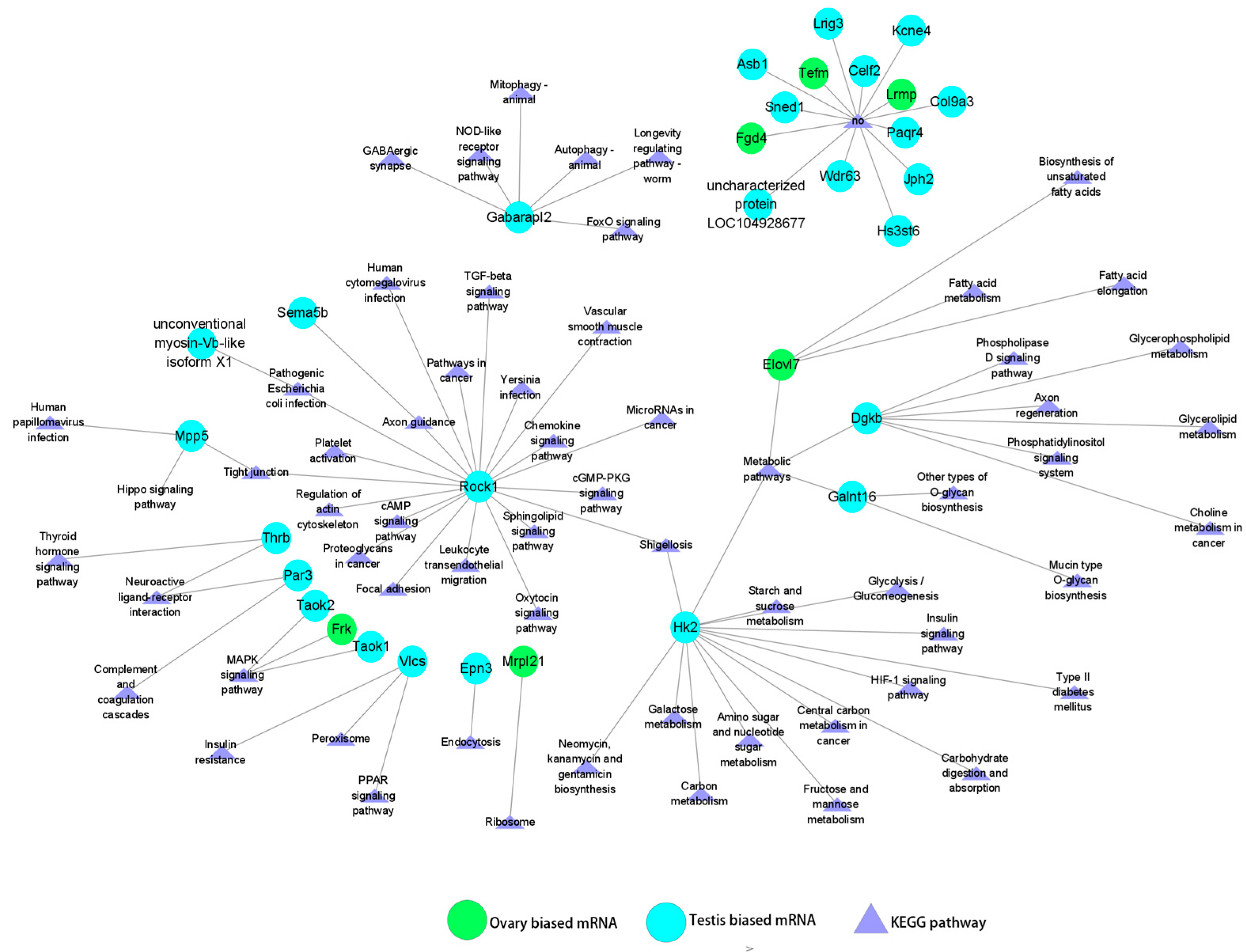
3.5. Regulatory Relationships Between miRNAs (DEMs) and Sex-Biased Genes (DEGs)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baroiller, J.F.; D’Cotta, H. The reversible sex of gonochoristic fish: Insights and consequences. Sex. Dev. 2016, 10, 242–266. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, Y.; Chakraborty, T.; Paul-Prasanth, B.; Ohta, K.; Nakamura, M. Sex determination, gonadal sex differentiation, and plasticity in vertebrate species. Physiol. Rev. 2021, 101, 1237–1308. [Google Scholar] [CrossRef]
- Hattori, R.S.; Strüssmann, C.A.; Fernandino, J.I.; Somoza, G.M. Genotypic sex determination in teleosts: Insights from the testis-determining amhy gene. Gen. Comp. Endocrinol. 2013, 192, 55–59. [Google Scholar] [CrossRef]
- Matsuda, M.; Nagahama, Y.; Shinomiya, A.; Sato, T.; Matsuda, C.; Kobayashi, T.; Morrey, C.E.; Shibata, N.; Asakawa, S.; Shimizu, N.; et al. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature 2002, 417, 559–563. [Google Scholar] [CrossRef]
- Liew, W.C.; Orbán, L. Zebrafish sex: A complicated affair. Brief. Funct. Genom. 2014, 13, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Du, X.; Chen, X.; Liu, L.; Wang, X. Transforming growth factor-β (TGF-β): A master signal pathway in teleost sex determination. Gen. Comp. Endocrinol. 2024, 355, 114561. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Jiang, J.; Wang, X.; Bártfai, R.; Kwan, H.Y.; Christoffels, A.; Orbán, L. Gonad differentiation in zebrafish is regulated by the canonical Wnt signaling pathway. Biol. Reprod. 2014, 90, 45. [Google Scholar] [CrossRef] [PubMed]
- Hattori, R.S.; Murai, Y.; Oura, M.; Masuda, S.; Majhi, S.K.; Sakamoto, T.; Fernandino, J.I.; Somoza, G.M.; Yokota, M.; Strüssmann, C.A. A Y-linked anti-Müllerian hormone duplication takes over a critical role in sex determination. Proc. Natl. Acad. Sci. USA 2012, 109, 2955–2959. [Google Scholar] [CrossRef]
- Jiang, D.N.; Yang, H.H.; Li, M.H.; Shi, H.J.; Zhang, X.B.; Wang, D.S. gsdf is a downstream gene of dmrt1 that functions in the male sex determination pathway of the Nile tilapia. Mol. Reprod. Dev. 2016, 83, 497–508. [Google Scholar] [CrossRef]
- Dai, S.; Qi, S.; Wei, X.; Liu, X.; Li, Y.; Zhou, X.; Xiao, H.; Lu, B.; Wang, D.; Li, M. Germline sexual fate is determined by the antagonistic action of dmrt1 and foxl3/foxl2 in tilapia. Development 2021, 148, dev199380. [Google Scholar] [CrossRef]
- Liu, X.; Dai, S.; Wu, J.; Wei, X.; Zhou, X.; Chen, M.; Tan, D.; Pu, D.; Li, M.; Wang, D. Roles of anti-Müllerian hormone and its duplicates in sex determination and germ cell proliferation of Nile tilapia. Genetics 2022, 220, iyab237. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Saitoh, N. Nuclear long non-coding RNAs as epigenetic regulators in cancer. Curr. Med. Chem. 2021, 28, 5098–5109. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Chen, K.; Jiang, M.; Jia, S.; Chen, J.; Tao, B.; Song, Y.; Li, Y.; Wang, Y.; Xiao, W.; et al. MiR-153b-3p regulates the proliferation and differentiation of male germ cells by targeting amh in common carp (Cyprinus carpio). Aquaculture 2021, 535, 736420. [Google Scholar] [CrossRef]
- Shen, X.; Yan, H.; Hu, M.; Zhou, H.; Wang, J.; Gao, R.; Liu, Q.; Wang, X.; Liu, Y. The potential regulatory role of the non-coding RNAs in regulating the exogenous estrogen-induced feminization in Takifugu rubripes gonad. Aquat. Toxicol. 2024, 273, 107022. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Meijer, H.A.; Kong, Y.W.; Lu, W.T.; Wilczynska, A.; Spriggs, R.V.; Robinson, S.W.; Godfrey, J.D.; Willis, A.E.; Bushell, M. Translational repression and eIF4A2 activity are critical for microRNA-mediated gene regulation. Science 2013, 340, 82–85. [Google Scholar] [CrossRef]
- Gu, W.; Xu, Y.; Xie, X.; Wang, T.; Ko, J.H.; Zhou, T. The role of RNA structure at 5′ untranslated region in microRNA-mediated gene regulation. RNA 2014, 20, 1369–1375. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Hurtado, A.; Mota-Gómez, I.; Lao, M.; Real, F.M.; Jedamzick, J.; Burgos, M.; Lupiáñez, D.G.; Jiménez, R.; Barrionuevo, F.J. Complete male-to-female sex reversal in XY mice lacking the miR-17~92 cluster. Nat. Commun. 2024, 15, 3809. [Google Scholar] [CrossRef]
- Peng, W.; Yu, S.; Handler, A.M.; Tu, Z.; Saccone, G.; Xi, Z.; Zhang, H. miRNA-1-3p is an early embryonic male sex-determining factor in the Oriental fruit fly Bactrocera dorsalis. Nat. Commun. 2020, 11, 932. [Google Scholar] [CrossRef]
- Yu, Q.; Peng, C.; Ye, Z.; Tang, Z.; Li, S.; Xiao, L.; Liu, S.; Yang, Y.; Zhao, M.; Zhang, Y.; et al. An estradiol-17β/miRNA-26a/cyp19a1a regulatory feedback loop in the protogynous hermaphroditic fish, Epinephelus coioides. Mol. Cell. Endocrinol. 2020, 504, 110689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Q.; Xu, W.; Wu, Z.; Li, D. Integrated analysis of miR-430 on steroidogenesis-related gene expression of larval rice field eel Monopterus albus. Int. J. Mol. Sci. 2021, 22, 6994. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; You, W.; Wang, Q.; Huang, F.; Shao, C. MicroRNA ssa-mir-196a-4 decreases lgr8 expression in testis development of Chinese tongue sole (Cynoglossus semilaevis). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2022, 258, 110695. [Google Scholar] [CrossRef]
- Zhao, L.; Dong, Q.; Luo, C.; Wu, Y.; Bu, D.; Qi, X.; Luo, Y.; Zhao, Y. DeepOmix: A scalable and interpretable multi-omics deep learning framework and application in cancer survival analysis. Comput. Struct. Biotechnol. J. 2021, 19, 2719–2725. [Google Scholar] [CrossRef]
- O’Connor, L.M.; O’Connor, B.A.; Lim, S.B.; Zeng, J.; Lo, C.H. Integrative multi-omics and systems bioinformatics in translational neuroscience: A data mining perspective. J. Pharm. Anal. 2023, 13, 836–850. [Google Scholar] [CrossRef] [PubMed]
- Skaftnesmo, K.O.; Edvardsen, R.B.; Furmanek, T.; Crespo, D.; Andersson, E.; Kleppe, L.; Taranger, G.L.; Bogerd, J.; Schulz, R.W.; Wargelius, A. Integrative testis transcriptome analysis reveals differentially expressed miRNAs and their mRNA targets during early puberty in Atlantic salmon. BMC Genom. 2017, 18, 801. [Google Scholar] [CrossRef]
- Liu, S.; Yang, Q.; Chen, Y.; Liu, Q.; Wang, W.; Song, J.; Zheng, Y.; Liu, W. Integrated analysis of mRNA- and miRNA-Seq in the ovary of rare minnow Gobiocypris rarus in response to 17α-methyltestosterone. Front. Genet. 2021, 12, 695699. [Google Scholar] [CrossRef]
- Lai, K.P.; Tam, N.Y.K.; Chen, Y.; Leung, C.T.; Lin, X.; Tsang, C.F.; Kwok, Y.C.; Tse, W.K.F.; Cheng, S.H.; Chan, T.F.; et al. miRNA-mRNA integrative analysis reveals the roles of miRNAs in hypoxia-altered embryonic development- and sex determination-related genes of medaka fish. Front. Mar. Sci. 2021, 8, 736362. [Google Scholar] [CrossRef]
- Assan, D.; Wang, Y.; Mustapha, U.F.; Ndandala, C.B.; Li, Z.; Li, G.L.; Chen, H. Neuropeptide Y in spotted scat (Scatophagus argus), characterization and functional analysis towards feed intake regulation. Fishes 2022, 7, 111. [Google Scholar] [CrossRef]
- Chen, H.; Jiang, D.; Li, Z.; Wang, Y.; Yang, X.; Li, S.; Li, S.; Yang, W.; Li, G. Comparative Physiological and Transcriptomic profiling offers insight into the sexual dimorphism of hepatic metabolism in size-dimorphic spotted Scat (Scatophagus argus). Life 2021, 11, 589. [Google Scholar] [CrossRef]
- Mustapha, U.F.; Huang, Y.; Huang, Y.Q.; Assan, D.; Shi, H.J.; Jiang, M.Y.; Deng, S.P.; Li, G.L.; Jiang, D.N. Gonadal development and molecular analysis revealed the critical window for sex differentiation, and E2 reversibility of XY-male spotted scat, Scatophagus argus. Aquaculture 2021, 544, 737147. [Google Scholar] [CrossRef]
- Huang, Y.-Q.; Zhang, X.-H.; Bian, C.; Jiao, K.-Z.; Zhang, L.; Huang, Y.; Yang, W.; Li, Y.; Shi, G.; Huang, Y.; et al. Allelic variation and duplication of the dmrt1 were associated with sex chromosome turnover in three representative Scatophagidae fish species. Commun. Biol. 2025, 8, 627. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc. 2008, 4, pdb-prot4986. [Google Scholar] [CrossRef]
- Jiao, K.; Li, Y.; Huang, Y.; Ndandala, C.B.; Shi, G.; Deng, S.; Shi, H.; Chen, H.; Li, G.; Jiang, D. Integrated analysis of the gonadal methylome and transcriptome provides new insights into the expression regulation of sex determination and differentiation genes in spotted scat (Scatophagus argus). Aquaculture 2024, 589, 740974. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- De Oliveira, L.F.; Christoff, A.P.; Margis, R. isomiRID: A framework to identify microRNA isoforms. Bioinformatics 2013, 29, 2521–2523. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2009, 26, 493–500. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 2005, 39, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Takahashi, T.; Ogiwara, K. cAMP signaling in ovarian physiology in teleosts: A review. Cell Signal 2023, 101, 110499. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Zhou, W.; Li, X.; Chen, H. The role of PKG in oocyte maturation of zebrafish. Biochem. Biophys. Res. Commun. 2018, 505, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qin, Y.; Schimenti, J.C. Gene regulation during meiosis. Trends Genet. 2024, 40, 326–336. [Google Scholar] [CrossRef]
- Xiao, S.; Cui, J.; Cao, Y.; Zhang, Y.; Yang, J.; Zheng, L.; Zhao, F.; Liu, X.; Zhou, Z.; Liu, D.; et al. Adolescent exposure to organophosphate insecticide malathion induces spermatogenesis dysfunction in mice by activating the HIF-1/MAPK/PI3K pathway. Environ. Pollut. 2024, 363, 125209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Jiang, H.; Ma, J.E.; Li, J.; Chen, J. Identification and differential expression of microRNAs in testis and ovary of Amur sturgeon (Acipenser schrenckii). Gene 2018, 658, 36–46. [Google Scholar] [CrossRef]
- He, P.; Wei, P.; Chen, X.; Lin, Y.; Peng, J. Identification and characterization of microRNAs in the gonad of Trachinotus ovatus using Solexa sequencing. Comp. Biochem. Physiol. Part D Genom. Proteom. 2019, 30, 312–320. [Google Scholar] [CrossRef]
- Shi, L.; Song, F.; Zhang, K.; Gu, Y.; Hu, J.; Sun, J.; Wang, Z.; Zhou, L.; Luo, J. Identification and Characterization of Sex-Biased miRNAs in the Golden Pompano (Trachinotus blochii). Animals 2022, 12, 3342. [Google Scholar] [CrossRef]
- Guo, K.; Liang, Z.; Li, F.; Wang, H. Comparison of miRNA and gene expression profiles between metastatic and primary prostate cancer. Oncol. Lett. 2017, 14, 6085–6090. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Ding, S.; Hao, Z.; Wang, X.; Zhan, Y.; Chang, Y. Integrated miRNA-mRNA analysis provides potential biomarkers for selective breeding in bay scallop (Argopecten irradians). Genomics 2021, 113, 2744–2755. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.F.; Chao, Q.H.; Huang, Q.Y.; Zhang, J.L. Expression of MIR-202-5P in gonads and verification of targeting relationship between MIR-202-5P and CBX 2 in Paralichthys olivaceus. Acta Hydrobiol. Sin. 2021, 45, 741–748. [Google Scholar] [CrossRef]
- Jiajie, T.; Yanzhou, Y.; Hoi-Hung, A.C.; Zi-Jiang, C.; Wai-Yee, C. Conserved miR-10 family represses proliferation and induces apoptosis in ovarian granulosa cells. Sci. Rep. 2017, 7, 41304. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, L.; Zhang, Y.; Madaniyati, M.; Shi, S.; Huang, L.; Song, X.; Pang, W.; Chu, G.; Yang, G. miR-10a-5p inhibits steroid hormone synthesis in porcine granulosa cells by targeting CREB1 and inhibiting cholesterol metabolism. Theriogenology 2023, 212, 19–29. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.; Zhang, X.; Lu, Y.; Li, L.; Cui, S. MiRNA-143 mediates the proliferative signaling pathway of FSH and regulates estradiol production. J. Endocrinol. 2017, 234, 1–14. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, C.Z.; Xu, M.Q.; Zhang, L.Q.; Liu, J.B.; Gao, Y.; Jiang, H.; Yuan, B.; Zhang, J.B. MiR-31 and miR-143 affect steroid hormone synthesis and inhibit cell apoptosis in bovine granulosa cells through FSHR. Theriogenology 2019, 123, 45–53. [Google Scholar] [CrossRef]
- Dai, A.; Sun, H.; Fang, T.; Zhang, Q.; Wu, S.; Jiang, Y.; Ding, L.; Yan, G.; Hu, Y. MicroRNA-133b stimulates ovarian estradiol synthesis by targeting Foxl2. FEBS Lett. 2013, 587, 2474–2482. [Google Scholar] [CrossRef]
- Tao, W.; Sun, L.; Shi, H.; Cheng, Y.; Jiang, D.; Fu, B.; Conte, M.A.; Gammerdinger, W.J.; Kocher, T.D.; Wang, D. Integrated analysis of miRNA and mRNA expression profiles in tilapia gonads at an early stage of sex differentiation. BMC Genom. 2016, 17, 328. [Google Scholar] [CrossRef]
- Li, Q.; Du, X.; Pan, Z.; Zhang, L.; Li, Q. The transcription factor SMAD4 and miR-10b contribute to E2 release and cell apoptosis in ovarian granulosa cells by targeting CYP19A1. Mol. Cell. Endocrinol. 2018, 476, 84–95. [Google Scholar] [CrossRef]
- Jiang, D.N.; Mustapha, U.F.; Shi, H.J.; Huang, Y.Q.; Si-Tu, J.X.; Wang, M.; Deng, S.P.; Chen, H.P.; Tian, C.X.; Zhu, C.H.; et al. Expression and transcriptional regulation of gsdf in spotted scat (Scatophagus argus). Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2019, 233, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.; Liu, Q.; Li, B.; An, L.; Hao, R.; Zhao, J.; Liu, S.; Song, J. Coordinated microRNA and messenger RNA expression profiles for understanding sexual dimorphism of gonads and the potential roles of microRNA in the steroidogenesis pathway in Nile tilapia (Oreochromis niloticus). Theriogenology 2016, 85, 970–978. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA | Primers (5′–3′) |
|---|---|
| novel_miR_1049 | GCTGTTCGGCTGAGCCTC |
| novel_miR_1203 | AAAGAGCACAGTGGCCTCCATAATC |
| novel_miR_1006 | TCTCTGATGCGGGCCATGACCAGTC |
| novel_miR_826 | TGCTGTGGTAGCCATGCTGGTT |
| sar-miR-10d-1 | TACCCTGTAGAACCGAATGTGT |
| novel_miR_579 | TCGTGTCTTGTGTTGCAGCCAGT |
| novel_miR_110 | TCCTCATTGTGCATGCTGTGTG |
| novel_miR_1112 | TGTGATATTGTTTCATGTGATTCG |
| Universal primer R | GATCGCCCTTCTACGTCGTAT |
| U6F | GCCACTTCGGCAGCACATAC |
| U6R | TTGCGTGTCATCCTTGC |
| Indexes | F1(%) | F2(%) | F3(%) | M1(%) | M2(%) | M3(%) |
|---|---|---|---|---|---|---|
| Raw reads | 18,797,843 | 11,972,637 | 11,847,428.5 | 9,939,298 | 11,847,428.5 | 12,599,932 |
| Clean reads | 18,690,270 (100%) | 11,935,960 (100%) | 11,774,117 (100%) | 9,888,937 (100%) | 11,795,830 (100%) | 12,518,068 (100%) |
| rRNA | 1,261,728 (6.75%) | 435,138 (3.65%) | 395,408 (3.36%) | 231,912 (2.35%) | 155,958 (1.32%) | 192,147 (1.53%) |
| snoRNA | 13,422 (0.07%) | 8637 (0.07%) | 1671 (0.01%) | 1100 (0.01%) | 1087 (0.01%) | 1456 (0.01%) |
| tRNA | 103,138 (0.55%) | 71,488 (0.60%) | 38,397 (0.33%) | 92,592 (0.94%) | 179,553 (1.52%) | 139,963 (1.12%) |
| Repbase | 248,340 (1.33%) | 164,456 (1.38%) | 123,269 (1.05%) | 27,764 (0.28%) | 30,414 (0.26%) | 36,912 (0.29%) |
| Unannotated | 17,063,642 (91.30%) | 11,256,241 (94.30%) | 11,215,372 (95.25%) | 9,535,569 (96.42%) | 11,428,818 (96.89%) | 12,147,590 (97.05%) |
| Mapped reads | 12,561,162 | 8,134,819 | 8,376,643 | 6,582,447 | 7,773,622 | 8,573,809 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, Y.; Jiao, K.; Huang, Y.; Wu, Y.; Shi, G.; Shi, H.; Chen, H.; Deng, S.; Li, G.; Tao, W.; et al. Integrated Analysis of Differential Expression Profiles of miRNA and mRNA in Gonads of Scatophagus argus Provides New Insights into Sexually Biased Gene Expression. Animals 2025, 15, 1564. https://doi.org/10.3390/ani15111564
Lei Y, Jiao K, Huang Y, Wu Y, Shi G, Shi H, Chen H, Deng S, Li G, Tao W, et al. Integrated Analysis of Differential Expression Profiles of miRNA and mRNA in Gonads of Scatophagus argus Provides New Insights into Sexually Biased Gene Expression. Animals. 2025; 15(11):1564. https://doi.org/10.3390/ani15111564
Chicago/Turabian StyleLei, Yaling, Kaizhi Jiao, Yuanqing Huang, Yuwei Wu, Gang Shi, Hongjuan Shi, Huapu Chen, Siping Deng, Guangli Li, Wenjing Tao, and et al. 2025. "Integrated Analysis of Differential Expression Profiles of miRNA and mRNA in Gonads of Scatophagus argus Provides New Insights into Sexually Biased Gene Expression" Animals 15, no. 11: 1564. https://doi.org/10.3390/ani15111564
APA StyleLei, Y., Jiao, K., Huang, Y., Wu, Y., Shi, G., Shi, H., Chen, H., Deng, S., Li, G., Tao, W., & Jiang, D. (2025). Integrated Analysis of Differential Expression Profiles of miRNA and mRNA in Gonads of Scatophagus argus Provides New Insights into Sexually Biased Gene Expression. Animals, 15(11), 1564. https://doi.org/10.3390/ani15111564