
Seasonal and Environmental Influences on the Gut Microbiota of South China Tigers (Panthera tigris amoyensis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Data Processing
2.4. Statistical Analyses
3. Results
3.1. Bacterial and Fungal Communities in the Gut of South China Tigers
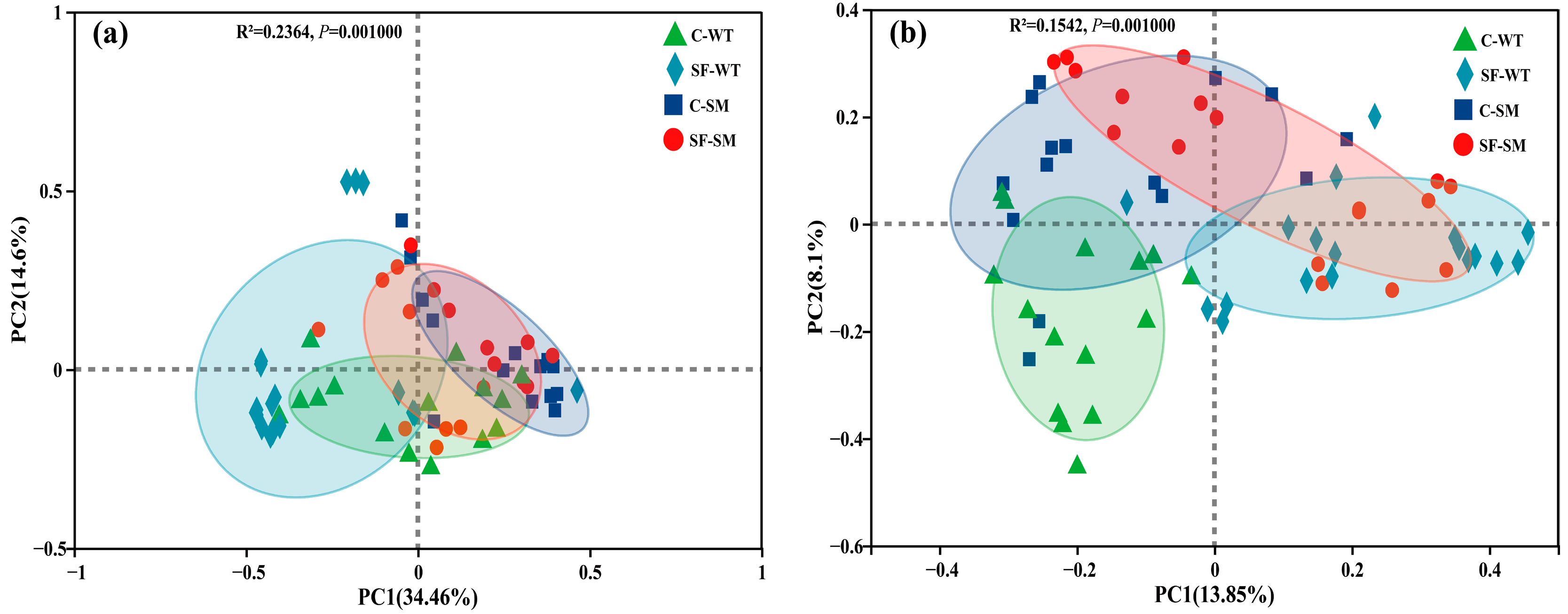
3.2. Effects of Environment and Seasonal Changes on Bacterial and Fungal Diversity
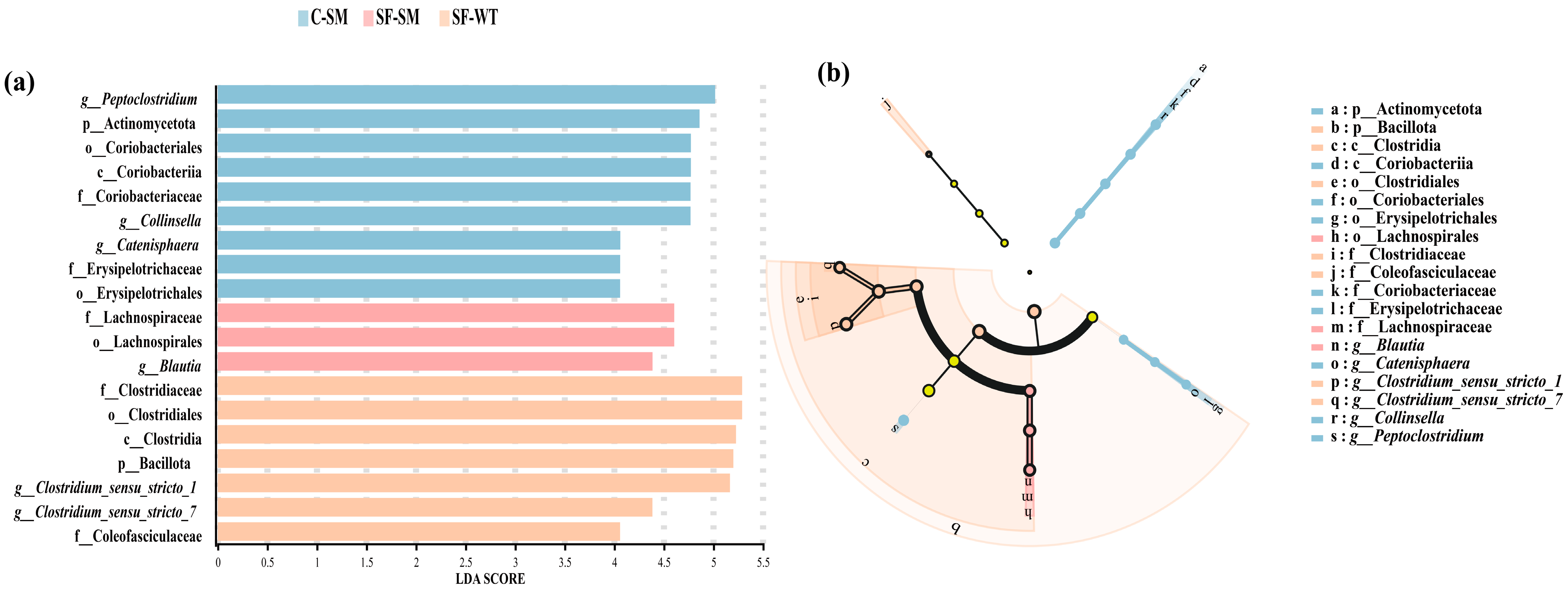
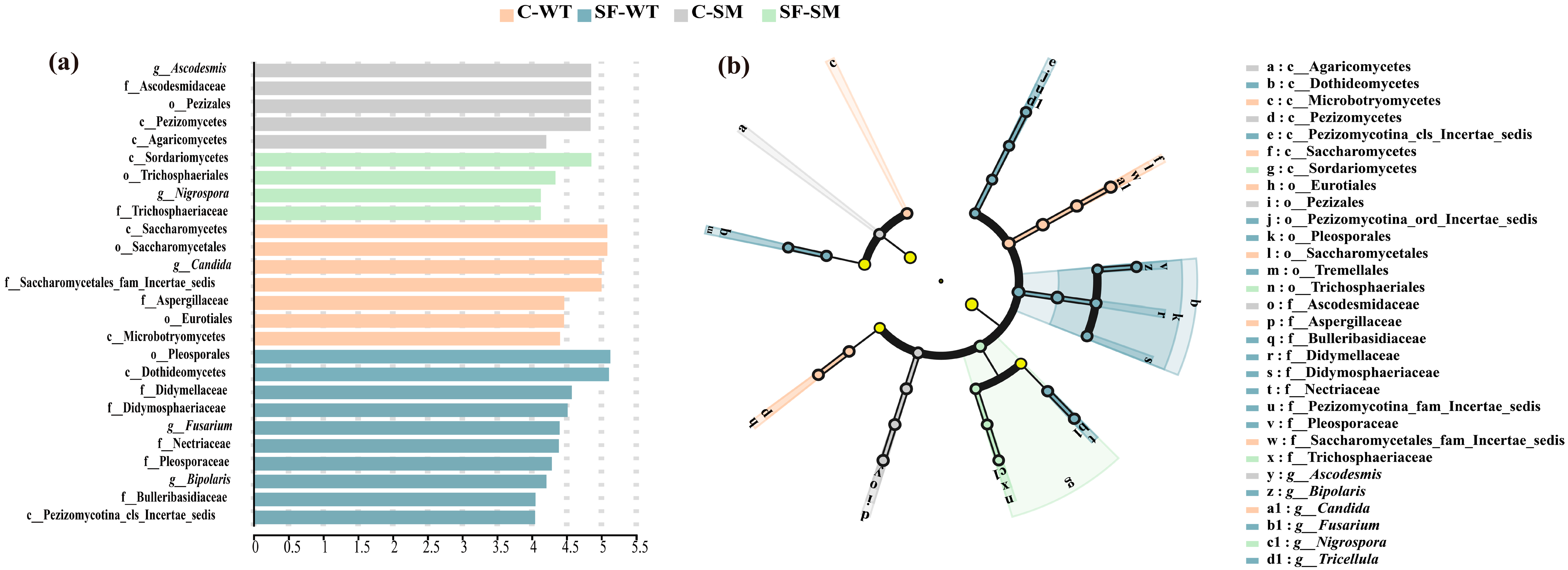
3.3. Effects of Environment and Seasonal Changes on the Dominant Gut Microbiota of South China Tigers
3.4. Prediction of Bacterial and Fungal Functions in the Gut of South China Tigers
4. Discussion
4.1. Dynamics and Diversity of Intestinal Bacterial Communities in South China Tigers
4.2. Intestinal Fungal Community Structure and Its Changes in South China Tigers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jepson, P.; Barua, M. A Theory of Flagship Species Action. Conserv. Soc. 2015, 13, 95–104. [Google Scholar] [CrossRef]
- Luo, Y.Q.; Xu, J.; Zhang, X.Y.; Hou, Y.L. Predicting the Impact of Climate Change on the Selection of Reintroduction Sites for the South China Tiger (Panthera tigris amoyensis) in China. Animals 2024, 14, 2477. [Google Scholar] [CrossRef] [PubMed]
- Ripple, W.J.; Estes, J.A.; Beschta, R.L.; Wilmers, C.C.; Ritchie, E.G.; Hebblewhite, M.; Berger, J.; Elmhagen, B.; Letnic, M.; Nelson, M.P. Status and ecological effects of the world’s largest carnivores. Science 2014, 343, 1241484. [Google Scholar] [CrossRef]
- Kang, A.; Xie, Y.; Tang, J.R.; Sanderson, E.W.; Ginsberg, J.R.; Zhang, E. Historic distribution and recent loss of tigers in China. Integr. Zool. 2010, 5, 335–341. [Google Scholar] [CrossRef]
- Goodrich, J.; Wibisono, H.; Miquelle, D.; Lynam, A.J.; Sanderson, E.; Chapman, S.; Gray, T.; Chanchani, P.; Harihar, A. Panthera tigris. The IUCN Red List of Threatened Species 2022. Available online: https://www.iucnredlist.org/species/15955/214862019 (accessed on 29 March 2025).
- Zhang, W.P.; Lin, K.X.; Fu, W.Y.; Xie, J.J.; Fan, X.Y.; Zhang, M.C.; Luo, H.X.; Yin, Y.Z.; Guo, Q.; Huang, H. Insights for the Captive Management of South China Tigers Based on a Large-Scale Genetic Survey. Genes 2024, 15, 398. [Google Scholar] [CrossRef]
- Luo, S.J.; Kim, J.H.; Johnson, W.E.; van der Welt, J.; Martenson, J.; Yuhki, N.; Miquelle, D.G.; Uphyrkina, O.; Goodrich, J.M.; Quigley, H.B. Phylogeography and genetic ancestry of tigers (Panthera tigris). PLoS Biol. 2004, 2, 442. [Google Scholar] [CrossRef]
- Huang, G.P.; Qi, D.W.; Yang, Z.S.; Hou, R.; Shi, W.Y.; Zhao, F.Q.; Li, Z.T.; Yan, L.; Wei, F.W. Gut microbiome as a key monitoring indicator for reintroductions of captive animals. Conserv. Biol. 2024, 38, e14173. [Google Scholar] [CrossRef]
- Curras, M.R.; Romanski, M.C.; Pauli, J.N. The pulsed effects of reintroducing wolves on the carnivore community of Isle Royale. Front. Ecol. Environ. 2024, 22, e2750. [Google Scholar]
- Zhang, W.P.; Xu, X.; Yue, B.S.; Hou, R.; Xie, J.J.; Zou, Z.T.; Han, Y.; Shen, F.J.; Zhang, L.; Xie, Z. Sorting out the genetic background of the last surviving South China tigers. J. Hered. 2019, 110, 641–650. [Google Scholar] [CrossRef]
- Shi, J.; Tian, C.; Xu, J.; Zhang, X.; Li, L.; Zhang, S. Applications of mitochondrial genes to an analysis of the phylogenetic relationship among tigers. J. South China Agric. Univ. 2014, 35, 13–17, 46. [Google Scholar]
- Wang, C.; Wu, D.D.; Yuan, Y.H.; Yao, M.C.; Han, J.L.; Wu, Y.J.; Shan, F.; Li, W.P.; Zhai, J.Q.; Huang, M.; et al. Population genomic analysis provides evidence of the past success and future potential of South China tiger captive conservation. BMC Biol. 2023, 21, 64. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.H.; Pei, E.; Liu, Q.X. Reproductive parameters of female South China Tigers in captivity. Eur. J. Wildl. Res. 2020, 66, 37. [Google Scholar] [CrossRef]
- Liu, H.M.; Yan, Q.; Zhao, B.; Luo, J.; Wang, C.M.; Du, Y.C.; Yan, J.; He, H.X. Isolation, molecular characterization, and phylogenetic analysis of encephalomyocarditis virus from South China tigers in China. Infect. Genet. Evol. 2013, 19, 240–243. [Google Scholar] [CrossRef]
- Chiu, H.C.; Fan, K.W.; Sun, X.S.; Lin, K.X.; Chen, T.T.; Yang, F.; Qiu, Y.F.; Wei, D.X.; Huang, C.Q. Detection and molecular characterisation of intestinal parasites in the South China tiger Panthera tigris amoyensis (Hilzheimer). Folia Parasitol. 2021, 68, 1–5. [Google Scholar] [CrossRef]
- Chiu, H.C.; Sun, X.S.; Bao, Y.L.; Fu, W.Y.; Lin, K.X.; Chen, T.T.; Zheng, C.Y.; Li, S.X.; Chen, W.T.; Huang, C.Q. Molecular identification of Colpodella sp. of South China tiger Panthera tigris amoyensis (Hilzheimer) in the Meihua Mountains, Fujian, China. Folia Parasitol. 2022, 69, 019. [Google Scholar] [CrossRef]
- Yuan, Y.H.; Wu, H.L.; Pan, H.J. Comparative analysis of the fecal microbiome and metabolomics of healthy versus captive South China tigers with mild diarrhea. Can. J. Microbiol. 2022, 68, 758–768. [Google Scholar] [CrossRef]
- Zhang, X.F.; Liao, Y.X.; Qin, T.; Ma, J.H.; Liu, J.X.; Zou, J.Q.; Huang, H.J.; Zhong, X.J.; Yang, M.H. Developmental stage variation in the gut microbiome of South China tigers. Front. Microbiol. 2022, 13, 962614. [Google Scholar] [CrossRef]
- Sun, Y.F.; Zhang, M.; Chen, T.T.; Fu, W.Y.; Xu, W.H.; Wu, Q.; Li, Y.; Zhu, Y.T.; Zhang, X.M.; Liu, L.Y.; et al. Comparative Study of Intestinal Microbial Community Diversity Between Captive South China Tiger (Panthera tigris amoyensis) Cubs and Adults at Meihuashan, Fujian, China. J. Wildl. 2020, 41, 912–918. (In Chinese) [Google Scholar]
- Sun, Y.F.; Yao, J.; Zhang, M.; Chen, T.T.; Xu, W.H.; Fu, W.Y.; Wu, Q.; Li, Y.; Chen, X.X.; Zhu, Y.T.; et al. Colonization and Development of the Fecal Microflora of South China Tiger Cubs (Panthera tigris amoyensis) by Sequencing of the 16S rRNA Gene. Microb. Physiol. 2022, 32, 18–29. [Google Scholar] [CrossRef]
- Ning, Y.; Qi, J.; Dobbins, M.T.; Liang, X.; Wang, J.; Chen, S.; Ma, J.; Jiang, G. Comparative Analysis of Microbial Community Structure and Function in the Gut of Wild and Captive Amur Tiger. Front. Microbiol. 2020, 11, 1665. [Google Scholar] [CrossRef]
- Williams, C.E.; Brown, A.E.; Williams, C.L. The role of diet and host species in shaping the seasonal dynamics of the gut microbiome. FEMS Microbiol. Ecol. 2023, 99, fiad156. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Pi, G.F.; Li, F. The role of intestinal flora in the regulation of bone homeostasis. Front. Cell. Infect. Microbiol. 2021, 11, 579323. [Google Scholar] [CrossRef] [PubMed]
- Huseyin, C.E.; O’Toole, P.W.; Cotter, P.D.; Scanlan, P.D. Forgotten fungi-the gut mycobiome in human health and disease. FEMS Microbiol. Rev. 2017, 41, 479–511. [Google Scholar] [CrossRef]
- Chin, V.K.; Yong, V.C.; Chong, P.P.; Nordin, S.A.; Basir, R.; Abdullah, M. Mycobiome in the Gut: A Multiperspective Review. Mediators Inflamm. 2020, 2020, 9560684. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Xiao, H.W.; Liu, X.Z. Research progress of gut mycobiota. Mycosystema 2023, 42, 26–37. [Google Scholar]
- Jiang, H.Y.; Chen, W.; Su, L.; Huang, M.W.; Lin, L.B.; Su, Q.; Li, G.Y.; Ahmad, H.I.; Li, L.M.; Zhang, X.J.; et al. Impact of host intraspecies genetic variation, diet, and age on bacterial and fungal intestinal microbiota in tigers. MicrobiologyOpen 2020, 9, e1050. [Google Scholar] [CrossRef]
- Wang, L.J.; Zhang, K.; Zeng, Y.J.; Luo, Y.T.; Peng, J.Y.; Zhang, J.; Kuang, T.T.; Fan, G. Gut mycobiome and metabolic diseases: The known, the unknown, and the future. Pharmacol. Res. 2023, 193, 106807. [Google Scholar] [CrossRef]
- Liu, C.S.; Zhao, D.F.; Ma, W.J.; Guo, Y.D.; Wang, A.J.; Wang, Q.L.; Lee, D.J. Denitrifying sulfide removal process on high-salinity wastewaters in the presence of Halomonas sp. Appl. Microbiol. Biotechnol. 2016, 100, 1421–1426. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Wu, X.Y.; Xia, Y.Y.; He, F.; Zhu, C.R.; Ren, W.K. Intestinal mycobiota in health and diseases: From a disrupted equilibrium to clinical opportunities. Microbiome 2021, 9, 60. [Google Scholar] [CrossRef]
- Li, X.V.; Leonardi, I.; Iliev, I.D. Gut Mycobiota in Immunity and Inflammatory Disease. Immunity 2019, 50, 1365–1379. [Google Scholar] [CrossRef]
- Gani, M.; Mohd-Ridwan, A.; Sitam, F.T.; Kamarudin, Z.; Selamat, S.S.; Awang, N.M.Z.; Karuppannan, K.V.; Md-Zain, B.M. Habitat shapes the gut microbiome diversity of Malayan tigers (Panthera tigris jacksoni) as revealed through metabarcoding 16S rRNA profiling. World J. Microbiol. Biotechnol. 2024, 40, 111. [Google Scholar] [CrossRef]
- Jia, X.Y. Study on Diversity and Function of Intestinal Bacterial Flora from African Lions (Panthera leo) of Different Seasons. Master’s Thesis, Jilin Agricultural University, Jilin, China, 2017. (In Chinese). [Google Scholar]
- Hua, Y.; Cao, H.Q.; Wang, J.; He, F.P.; Jiang, G.S. Gut microbiota and fecal metabolites in captive and wild North China leopard (Panthera pardus japonensis) by comparsion using 16 s rRNA gene sequencing and LC/MS-based metabolomics. BMC Vet. Res. 2020, 16, 363. [Google Scholar] [CrossRef]
- Liu, M.; Zhu, J.; Cheng, J.X.; Ding, H.X.; Gao, Y.; Jiang, W.J.; Yuan, K.; Dong, X.; Chen, L. Characterization of Snow Leopard (Panthera uncia) Fecal Microbiota by Barcoded Pyrosequencing. Acta Theriol. Sin. 2019, 40, 873–881. (In Chinese) [Google Scholar]
- Dias, B.d.C.; Lamarca, A.P.; Machado, D.T.; Kloh, V.P.; de Carvalho, F.M.; Vasconcelos, A.T.R. Metabolic pathways associated with Firmicutes prevalence in the gut of multiple livestock animals and humans. Anim. Microb. 2025, 7, 20. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, S.; Nie, Q.; He, H.; Tan, H.; Geng, F.; Ji, H.; Hu, J.; Nie, S. Gut firmicutes: Relationship with dietary fiber and role in host homeostasis. Crit. Rev. Food Sci. Nutr. 2023, 63, 12073–12088. [Google Scholar] [CrossRef]
- Walsh, A.J.; Bryant, R.V.; Travis, S.P.L. Current best practice for disease activity assessment in IBD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Korobkina, E.D.; Calejman, C.M.; Haley, J.A.; Kelly, M.E.; Li, H.W.; Gaughan, M.; Chen, Q.B.; Pepper, H.L.; Ahmad, H.; Boucher, A.; et al. Brown fat ATP-citrate lyase links carbohydrate availability to thermogenesis and guards against metabolic stress. Nat. Metab. 2024, 6, 4717. [Google Scholar] [CrossRef] [PubMed]
- Khairulmunir, M.; Gani, M.; Mohd-Ridwan, A.R.; Karuppannan, K.V.; Abdul-Latiff, M.A.B.; Md-Zain, B.M. Alteration of the gut microbial composition of critically endangered Malayan tigers (Panthera tigris jacksoni) in captivity during enrichment phase. Mol. Biol. Rep. 2024, 51, 742. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Lang, P.F.; Wang, H.T.; Wang, J.; Cao, H.Q.; Jiang, G.S. Analysis of Intestinal Fungus Diversity of Wild and Captive North—Chinese Leopard (Panthera pardus japonensis) Based on High-Throughput Sequencing. Acta Theriol. Sin. 2020, 41, 5–14. (In Chinese) [Google Scholar]
- Mukherjee, P.K.; Sendid, B.; Hoarau, G.; Colombel, J.F.; Poulain, D.; Ghannoum, M.A. Mycobiota in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 77–87. [Google Scholar] [CrossRef]
- Richard, M.L.; Lamas, B.; Liguori, G.; Hoffmann, T.W.; Sokol, H. Gut Fungal Microbiota: The Yin and Yang of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 656–665. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Xu, X.; Zhang, Z.; Zhang, X.; Lin, K.; Luo, H.; Huang, C.; Lin, X.; Zhang, C.; Qing, Y.; et al. Seasonal and Environmental Influences on the Gut Microbiota of South China Tigers (Panthera tigris amoyensis). Animals 2025, 15, 1471. https://doi.org/10.3390/ani15101471
Zhou L, Xu X, Zhang Z, Zhang X, Lin K, Luo H, Huang C, Lin X, Zhang C, Qing Y, et al. Seasonal and Environmental Influences on the Gut Microbiota of South China Tigers (Panthera tigris amoyensis). Animals. 2025; 15(10):1471. https://doi.org/10.3390/ani15101471
Chicago/Turabian StyleZhou, Li, Xiyao Xu, Zhirong Zhang, Xu Zhang, Kaixiong Lin, Hongxing Luo, Cheng Huang, Xipan Lin, Chunli Zhang, Yan Qing, and et al. 2025. "Seasonal and Environmental Influences on the Gut Microbiota of South China Tigers (Panthera tigris amoyensis)" Animals 15, no. 10: 1471. https://doi.org/10.3390/ani15101471
APA StyleZhou, L., Xu, X., Zhang, Z., Zhang, X., Lin, K., Luo, H., Huang, C., Lin, X., Zhang, C., Qing, Y., Teng, L., & Liu, Z. (2025). Seasonal and Environmental Influences on the Gut Microbiota of South China Tigers (Panthera tigris amoyensis). Animals, 15(10), 1471. https://doi.org/10.3390/ani15101471