

Preliminary Study on the Genetic Structure and Functional Candidate Genes of Grassland-Thoroughbreds Based on Whole-Genome Resequencing
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Library Construction
2.3. Quality Control and Data Filtering
2.4. Variant Detection and Annotation
2.5. Population Structure Analysis
2.6. Detection of Selection Signatures
2.7. Functional Enrichment Analysis
3. Results
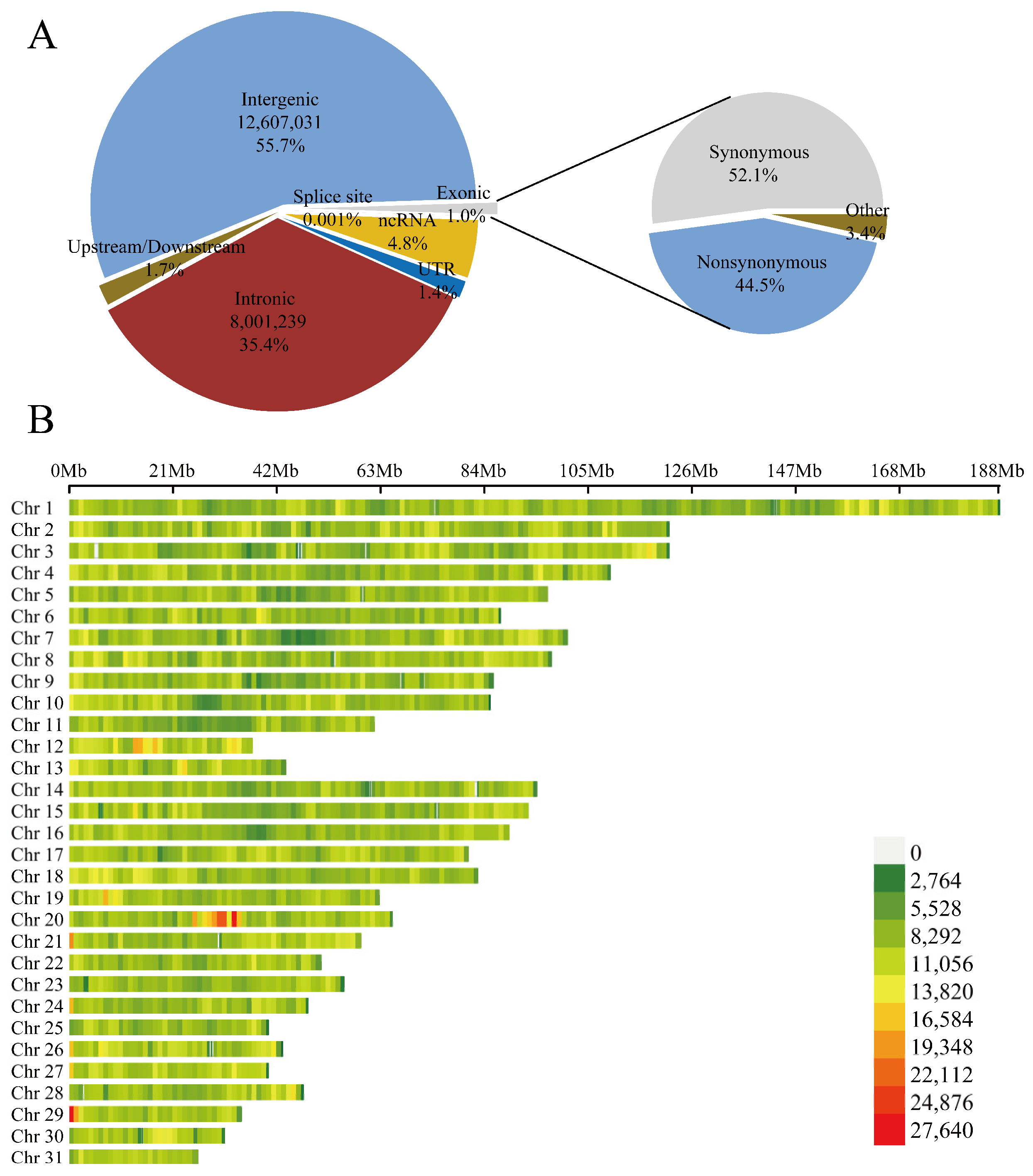
3.1. Sequencing Data and Genome-Wide Genetic Variation
3.2. Population Structure Analysis
3.3. Selection Signal Analysis and Functional Enrichment
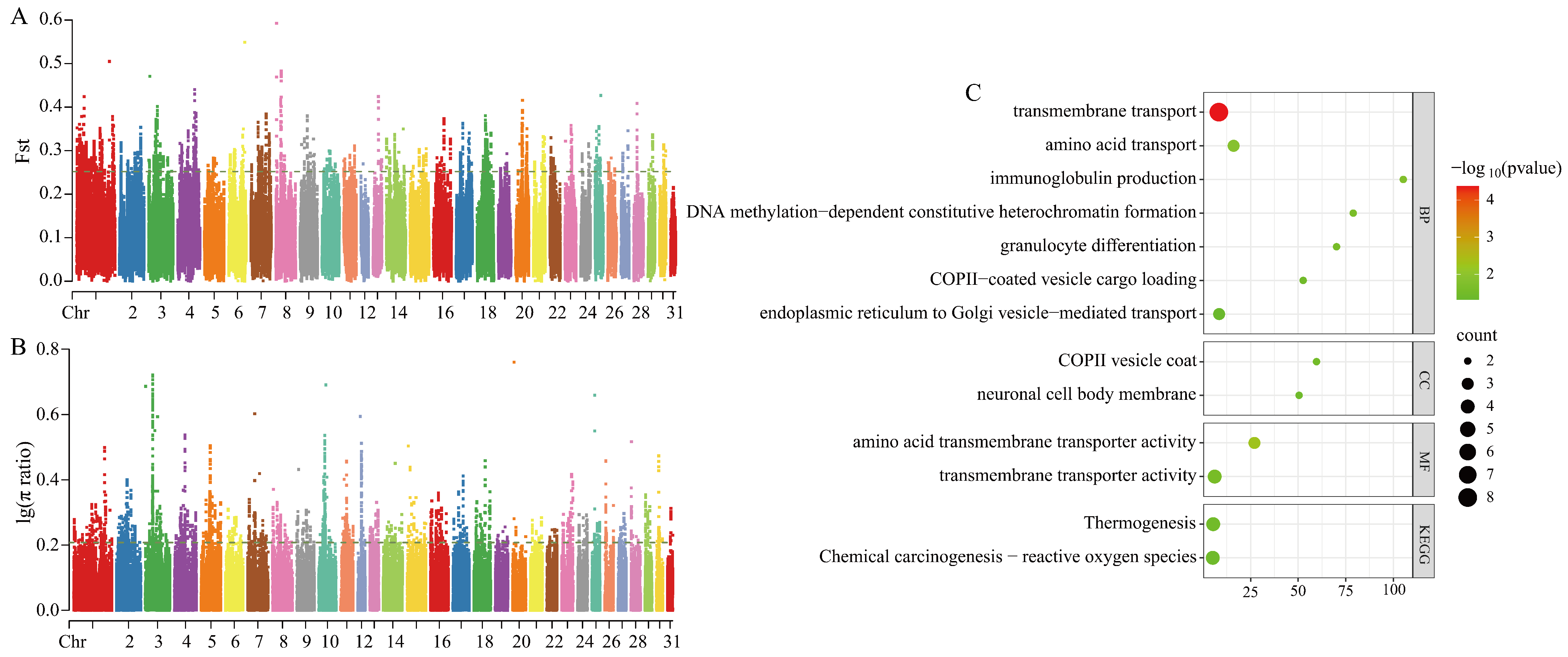
3.3.1. Selection Signal Detection Between Grassland-Thoroughbred and Mongolian Horse
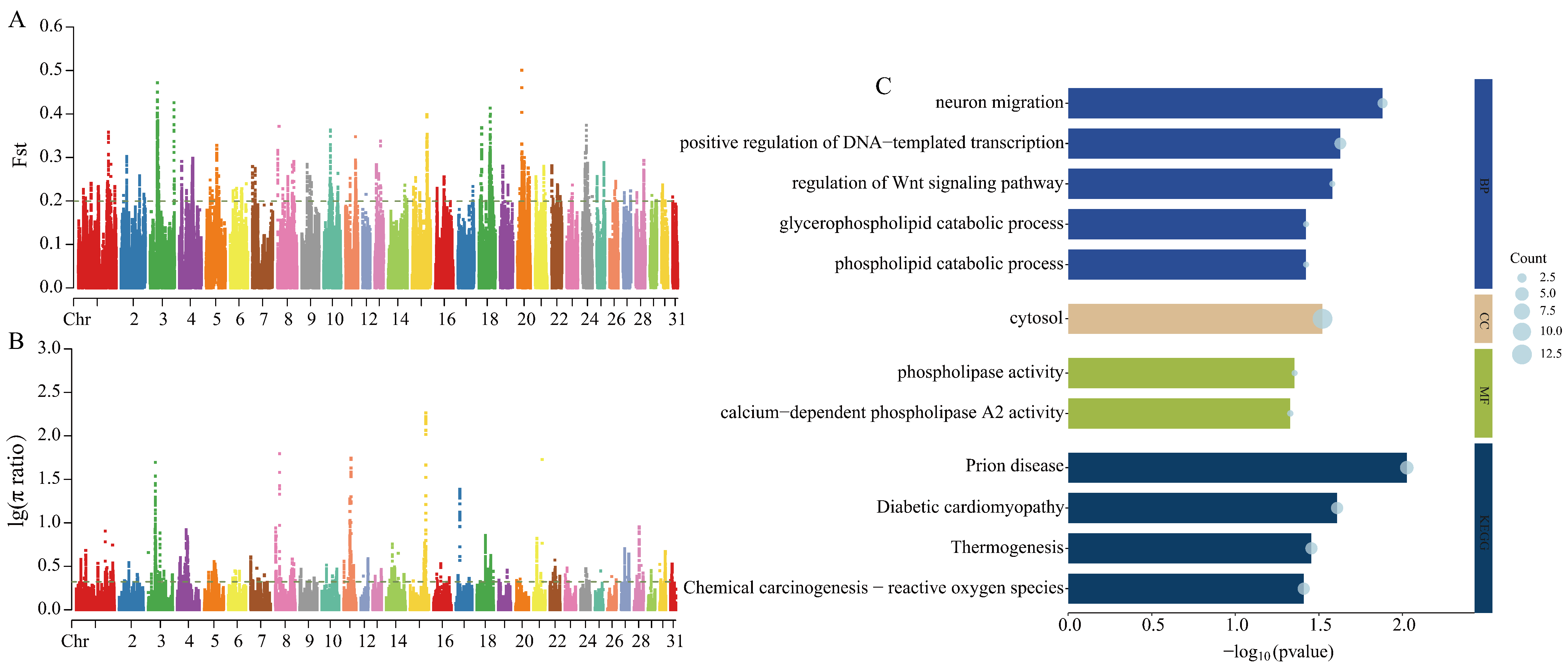
3.3.2. Selection Signal Detection Between Grassland-Thoroughbred and Thoroughbred Horse
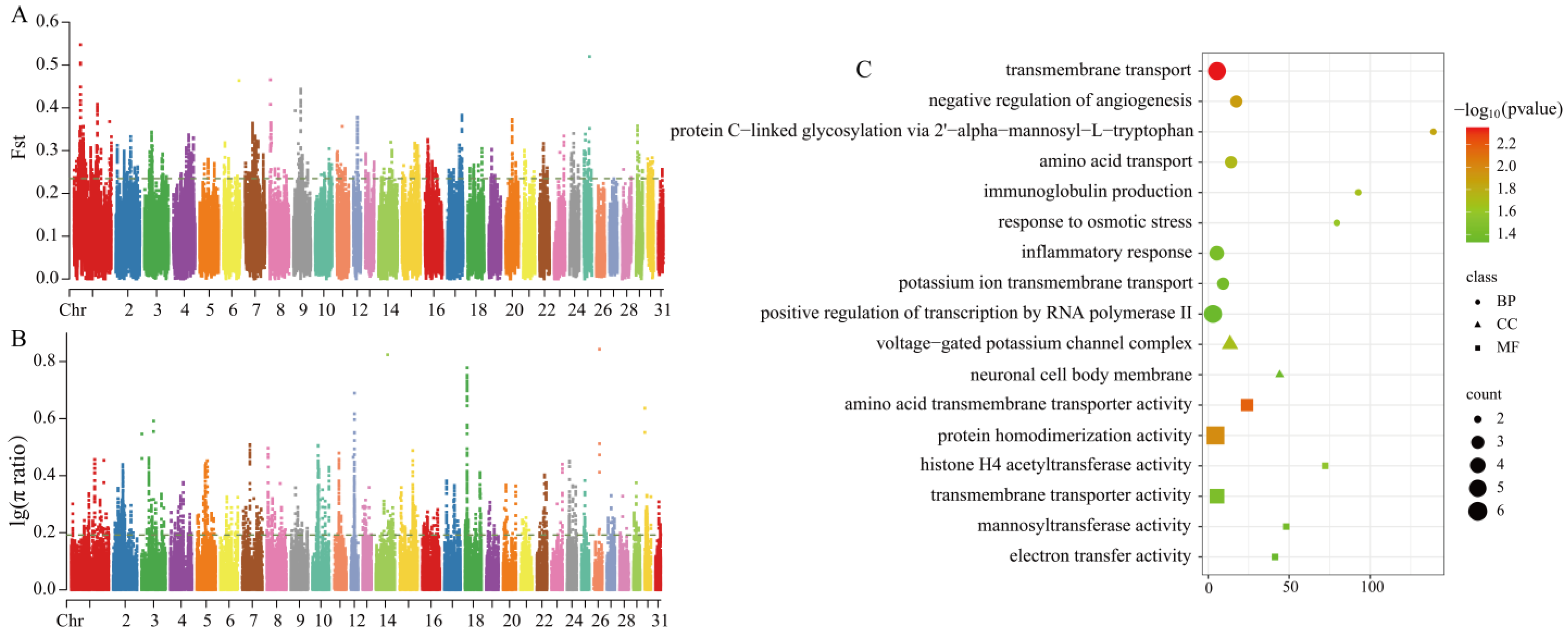
3.3.3. Selection Signal Detection Between Grassland-Thoroughbred and Xilingol Horse
3.3.4. Candidate Genes Under Selection in the Grassland-Thoroughbred
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moazemi, I.; Mohammadabadi, M.; Mostafavi, A.; Esmailizadeh, A.; Babenko, O.; Bushtruk, M.; Tkachenko, S.; Stavetska, R.; Klopenko, N. Polymorphism of DMRT3 gene and its association with body measurements in horse breeds. Russ. J. Genet. 2020, 56, 1232–1240. [Google Scholar] [CrossRef]
- International Federation of Horse Racing Authorities. IFHRA Annual Report 2019; International Federation of Horse Racing Authorities: Boulogne, France, 2019. [Google Scholar]
- Chien, P.M.; Council for Australian University Tourism and Hospitality Education. CAUTHE 2014: Tourism and Hospitality in the Contemporary World: Trends, Changes and Complexity: Trends, Changes and Complexity; School of Tourism, The University of Queensland: Brisbane, Australia, 2014. [Google Scholar]
- Worthington, A.C. National exuberance: A note on the Melbourne Cup effect in Australian stock returns. Econ. Pap. A J. Appl. Econ. Policy 2007, 26, 170–179. [Google Scholar] [CrossRef]
- Narayan, P.K.; Smyth, R. The race that stops a nation: The demand for the Melbourne Cup. Econ. Rec. 2004, 80, 193–207. [Google Scholar] [CrossRef]
- Davis, M. When Things Get Dark: A Mongolian Winter’s Tale; Macmillan: New York, NY, USA, 2010. [Google Scholar]
- Qi, B. Xilin Gol League Animal Husbandry Chronicle; Inner Mongolia People’s Publishing House: Hohhot, China, 2002. [Google Scholar]
- Cassidy, R. The Sport of Kings: Kinship, Class and Thoroughbred Breeding in Newmarket; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Kay, J.; Vamplew, W. Encyclopedia of British Horse Racing; Routledge: London, UK, 2012. [Google Scholar]
- Porto-Neto, L.; Lee, S.; Sonstegard, T.; Van Tassell, C.; Lee, H.; Gibson, J.; Gondro, C. Genome-wide detection of signatures of selection in K orean H anwoo cattle. Anim. Genet. 2014, 45, 180–190. [Google Scholar] [CrossRef]
- Zhong, X.; Hao, S.; Zhang, Z.; Zhao, Q.-B.; Olasege, B.S.; Qiu-Meng, L.; Yang, Y.; Zhang, X.-Z.; Wang, Q.-S.; Pan, Y.-C. Genome-wide detection of selective signatures in a Jinhua pig population. J. Integr. Agric. 2020, 19, 1314–1322. [Google Scholar]
- Bordbar, F.; Mohammadabadi, M.; Jensen, J.; Xu, L.; Li, J.; Zhang, L. Identification of candidate genes regulating carcass depth and hind leg circumference in simmental beef cattle using Illumina Bovine Beadchip and next-generation sequencing analyses. Animals 2022, 12, 1103. [Google Scholar] [CrossRef]
- Consortium, I.H. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.P. A map of human genome variation from population scale sequencing. Nature 2010, 467, 1061. [Google Scholar] [CrossRef]
- Bs, W. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Deniskova, T.E.; Yudin, N.S.; Dotsev, A.V.; Khamiruev, T.N.; Selionova, M.I.; Egorov, S.V.; Reyer, H.; Wimmers, K.; Brem, G.; et al. High-density genotyping reveals signatures of selection related to acclimation and economically important traits in 15 local sheep breeds from Russia. BMC Genom. 2019, 20, 294. [Google Scholar] [CrossRef]
- Wei, C.; Wang, H.; Liu, G.; Zhao, F.; Kijas, J.W.; Ma, Y.; Lu, J.; Zhang, L.; Cao, J.; Wu, M. Genome-wide analysis reveals adaptation to high altitudes in Tibetan sheep. Sci. Rep. 2016, 6, 26770. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Hendricks, B.L. International Encyclopedia of Horse Breeds; University of Oklahoma Press: Norman, OK, USA, 2007. [Google Scholar]
- Wadley, G.D.; McConell, G.K. High-dose antioxidant vitamin C supplementation does not prevent acute exercise-induced increases in markers of skeletal muscle mitochondrial biogenesis in rats. J. Appl. Physiol. 2010, 108, 1719–1726. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Pohnert, S.C.; Li, P.; Zhang, M.; Gumbs, C.; Rosenberg, P.B.; Williams, R.S.; Yan, Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J. Biol. Chem. 2005, 280, 19587–19593. [Google Scholar] [CrossRef]
- Chou, T.-J.; Lu, C.-W.; Lin, L.-Y.; Hsu, Y.-J.; Huang, C.-C.; Huang, K.-C. Proteomic analysis of skeletal muscle and white adipose tissue after aerobic exercise training in high fat diet induced obese mice. Int. J. Mol. Sci. 2023, 24, 5743. [Google Scholar] [CrossRef]
- Alizadeh, R.; Salehi, O.; Rezaeinezhad, N.; Hosseini, S.A. The effect of high intensity interval training with genistein supplementation on mitochondrial function in the heart tissue of elderly rats. Exp. Gerontol. 2023, 171, 112039. [Google Scholar] [CrossRef]
- Feil, R.; Hofmann, F.; Kleppisch, T. Function of cGMP-dependent protein kinases in the nervous system. Rev. Neurosci. 2005, 16, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Yao, W.; Zhou, R.; Pei, Z. Exercise training affects calcium ion transport by downregulating the CACNA2D1 protein to reduce hypertension-induced myocardial injury in mice. iScience 2024, 27, 109351. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Park, K.-D.; Taye, M.; Lee, C.; Kim, H.; Lee, H.-K.; Shin, D. Analysis of cross-population differentiation between Thoroughbred and Jeju horses. Asian-Australas. J. Anim. Sci. 2017, 31, 1110. [Google Scholar] [CrossRef] [PubMed]
- Perez, K.; Ciotlos, S.; McGirr, J.; Limbad, C.; Doi, R.; Nederveen, J.P.; Nilsson, M.I.; Winer, D.A.; Evans, W.; Tarnopolsky, M.; et al. Single nuclei profiling identifies cell specific markers of skeletal muscle aging, frailty, and senescence. Aging 2022, 14, 9393–9422. [Google Scholar] [CrossRef]
- Riedl, I.; Yoshioka, M.; Nishida, Y.; Tobina, T.; Paradis, R.; Shono, N.; Tanaka, H.; St-Amand, J. Regulation of skeletal muscle transcriptome in elderly men after 6 weeks of endurance training at lactate threshold intensity. Exp. Gerontol. 2010, 45, 896–903. [Google Scholar] [CrossRef]
- Mascher, H.; Andersson, H.; Nilsson, P.A.; Ekblom, B.; Blomstrand, E. Changes in signalling pathways regulating protein synthesis in human muscle in the recovery period after endurance exercise. Acta Physiol. 2007, 191, 67–75. [Google Scholar] [CrossRef]
- Léger, B.; Cartoni, R.; Praz, M.; Lamon, S.; Dériaz, O.; Crettenand, A.; Gobelet, C.; Rohmer, P.; Konzelmann, M.; Luthi, F. Akt signalling through GSK-3β, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2006, 576, 923–933. [Google Scholar] [CrossRef]
- Stebbings, G.; Williams, A.; Herbert, A.J.; Lockey, S.; Heffernan, S.; Erskine, R.; Morse, C.; Day, S. TTN genotype is associated with fascicle length and marathon running performance. Scand. J. Med. Sci. Sports 2018, 28, 400–406. [Google Scholar] [CrossRef]
- Ding, W.; Gong, W.; Bou, T.; Shi, L.; Lin, Y.; Wu, H.; Dugarjaviin, M.; Bai, D. Pilot Study on the Profiling and Functional Analysis of mRNA, miRNA, and lncRNA in the Skeletal Muscle of Mongolian Horses, Xilingol Horses, and Grassland-Thoroughbreds. Animals 2025, 15, 1123. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; von Maltzahn, J.; Dumont, N.A.; Stark, D.A.; Wang, Y.X.; Nhan, K.; Frenette, J.; Cornelison, D.; Rudnicki, M.A. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J. Cell Biol. 2014, 205, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Baker, J.; Cracknell, T.; Haynes, A.R.; Blanco, G. IGFN1_v1 is required for myoblast fusion and differentiation. PLoS ONE 2017, 12, e0180217. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, S.; Ojala, K.; Kallioniemi, O. Analysis of kinase gene expression patterns across 5681 human tissue samples reveals functional genomic taxonomy of the kinome. PLoS ONE 2010, 5, e15068. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, T.R. Revealing the Role of IGFN1 in Skeletal Muscle; University of York: York, UK, 2019. [Google Scholar]
- Wang, W.; Olson, D.; Liang, G.; Franceschi, R.T.; Li, C.; Wang, B.; Wang, S.S.; Yang, S. Collagen XXIV (Col24α1) promotes osteoblastic differentiation and mineralization through TGF-β/Smads signaling pathway. Int. J. Biol. Sci. 2012, 8, 1310. [Google Scholar] [CrossRef]
- Bustos, F.; de la Vega, E.; Cabezas, F.; Thompson, J.; Cornelison, D.D.; Olwin, B.B.; Yates, J.R., 3rd; Olguín, H.C. NEDD4 Regulates PAX7 Levels Promoting Activation of the Differentiation Program in Skeletal Muscle Precursors. Stem Cells 2015, 33, 3138–3151. [Google Scholar] [CrossRef]
- Wang, T.; Feng, S.; Zhou, H.; Mao, W.; Bai, R.; Xia, Y.; Huang, J.; Zhang, R.; Lin, F. PIEZO1 activation enhances myogenesis and mitigates muscle degeneration in rotator cuff tear. Regen. Ther. 2025, 28, 143–152. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, W.; Gong, W.; Bou, T.; Shi, L.; Lin, Y.; Shi, X.; Li, Z.; Wu, H.; Dugarjaviin, M.; Bai, D.; et al. Preliminary Study on the Genetic Structure and Functional Candidate Genes of Grassland-Thoroughbreds Based on Whole-Genome Resequencing. Animals 2025, 15, 1462. https://doi.org/10.3390/ani15101462
Ding W, Gong W, Bou T, Shi L, Lin Y, Shi X, Li Z, Wu H, Dugarjaviin M, Bai D, et al. Preliminary Study on the Genetic Structure and Functional Candidate Genes of Grassland-Thoroughbreds Based on Whole-Genome Resequencing. Animals. 2025; 15(10):1462. https://doi.org/10.3390/ani15101462
Chicago/Turabian StyleDing, Wenqi, Wendian Gong, Tugeqin Bou, Lin Shi, Yanan Lin, Xiaoyuan Shi, Zheng Li, Huize Wu, Manglai Dugarjaviin, Dongyi Bai, and et al. 2025. "Preliminary Study on the Genetic Structure and Functional Candidate Genes of Grassland-Thoroughbreds Based on Whole-Genome Resequencing" Animals 15, no. 10: 1462. https://doi.org/10.3390/ani15101462
APA StyleDing, W., Gong, W., Bou, T., Shi, L., Lin, Y., Shi, X., Li, Z., Wu, H., Dugarjaviin, M., Bai, D., & Zhao, Y. (2025). Preliminary Study on the Genetic Structure and Functional Candidate Genes of Grassland-Thoroughbreds Based on Whole-Genome Resequencing. Animals, 15(10), 1462. https://doi.org/10.3390/ani15101462