
Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies

 , , , , , , and
, , , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Background
2.2. Faecal Dropping Samples Collection
2.3. Cloacal Swab Samples Collection
2.4. Statistical Analysis
2.5. Sample Processing and Molecular Detection of AIVs
2.6. Virus Isolation in Specific Pathogen Free (SPF) Embryonated Eggs
2.7. Genome Sequencing and Phylogenetic Analysis
2.8. Sequence Analysis
2.9. Cytochrome Oxidase I (COI) Barcoding of Faecal Dropping Samples for Host Identification
3. Results
3.1. Wild Bird Population Sampled
3.2. AIVs Detection in Faecal Droppings and Cloacal Swabs
3.3. Sequence Analysis
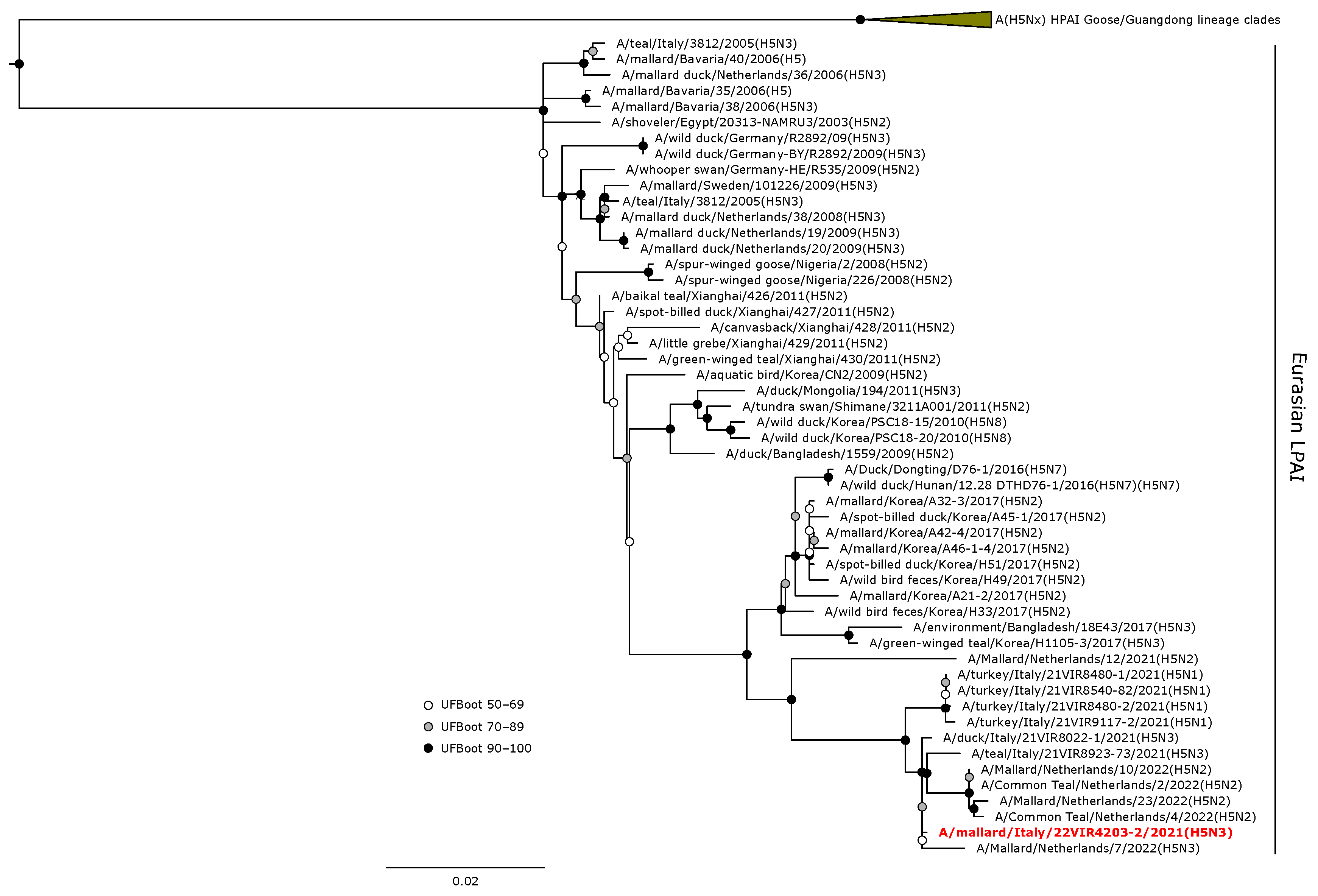
3.4. Phylogenetic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef]
- Horman, W.S.J.; Nguyen, T.H.O.; Kedzierska, K.; Bean, A.G.D.; Layton, D.S. The Drivers of Pathology in Zoonotic Avian Influenza: The Interplay Between Host and Pathogen. Front. Immunol. 2018, 9, 1812. [Google Scholar] [CrossRef] [PubMed]
- Stallknecht, D.E.; Brown, J.D. Ecology of Avian Influenza in Wild Birds. In Avian Influenza; Swayne, D.E., Ed.; Wiley-Blackwell: Ames, IA, USA, 2008; pp. 43–58. [Google Scholar]
- Bevins, S.; Shriner, S.; Cumbee, J.; Dilione, K.; Douglass, K.; Ellis, J.; Killian, M.L.; Torchetti, M.; Lenoch, J. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006. [Google Scholar] [CrossRef] [PubMed]
- Lo, F.T.; Zecchin, B.; Diallo, A.A.; Racky, O.; Tassoni, L.; Diop, A.; Diouf, M.; Diouf, M.; Samb, Y.N.; Pastori, A.; et al. Intercontinental Spread of Eurasian Highly Pathogenic Avian Influenza A(H5N1) to Senegal. Emerg. Infect. Dis. 2022, 28, 234–237. [Google Scholar] [CrossRef] [PubMed]
- The Global Consortium for H5N8 and Related Influenza Viruses. Role for migratory wild birds in the global spread of avian influenza H5N8. Science 2016, 354, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Pantin-Jackwood, M.J.; Swayne, D.E. Pathogenesis and pathobiology of avian influenza virus infection in birds. Rev. Sci. Tech. 2009, 28, 113–136. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.; Speidel, E.S.; Moore, E.; Plowright, L.; Piccirillo, A.; Capua, I.; Cordioli, P.; Fioretti, A.; Alexander, D.J. Changes in the haemagglutinin and the neuraminidase genes prior to the emergence of highly pathogenic H7N1 avian influenza viruses in Italy. Arch. Virol. 2001, 146, 963–973. [Google Scholar] [CrossRef]
- Röhm, C.; Horimoto, T.; Kawaoka, Y.; Süss, J.; Webster, R.G. Do hemagglutinin genes of highly pathogenic avian influenza viruses constitute unique phylogenetic lineages? Virology 1995, 209, 664–670. [Google Scholar] [CrossRef][Green Version]
- Xu, X.; Subbarao; Cox, N.J.; Guo, Y. Genetic characterization of the pathogenic influenza A/Goose/Guangdong/1/96 (H5N1) virus: Similarity of its hemagglutinin gene to those of H5N1 viruses from the 1997 outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef]
- Chen, H.; Smith, G.J.D.; Zhang, S.Y.; Qin, K.; Wang, J.; Li, K.S.; Webster, R.G.; Peiris, J.S.M.; Guan, Y. H5N1 virus outbreak in migratory waterfowl. Nature 2005, 436, 191–192. [Google Scholar] [CrossRef]
- Ellis, T.M.; Bousfield, R.B.; Bissett, L.A.; Dyrting, K.C.; Luk, G.S.; Tsim, S.T.; Sturm-Ramirez, K.; Webster, R.G.; Guan, Y.; Malik Peiris, J.S. Investigation of outbreaks of highly pathogenic H5N1 avian influenza in waterfowl and wild birds in Hong Kong in late 2002. Avian. Pathol. 2004, 33, 492–505. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Bahl, J.; Riley, S.; Duan, L.; Zhang, J.X.; Chen, H.; Peiris, J.S.M.; Smith, G.J.D.; Guan, Y. Evolutionary Dynamics and Emergence of Panzootic H5N1 Influenza Viruses. PLoS Pathog. 2008, 4, e1000161. [Google Scholar] [CrossRef]
- World Health Organization. Antigenic and genetic characteristics of zoonotic influenza viruses and development of candidate vaccine viruses for pandemic preparedness. Wkly. Epidemiol. Rec. 2018, 93, 142–152. [Google Scholar]
- Beerens, N.; Heutink, R.; Harders, F.; Roose, M.; Pritz-Verschuren, S.B.E.; Germeraad, E.A.; Engelsma, M. Incursion of Novel Highly Pathogenic Avian Influenza A(H5N8) Virus, the Netherlands, October 2020. Emerg. Infect. Dis. 2021, 27, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, E.; Staubach, C.; et al. Avian influenza overview September–December 2022. EFSA J. 2023, 21, e07786. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, E.; Staubach, C.; et al. Avian influenza overview December 2021–March 2022. EFSA J. 2022, 20, e07289. [Google Scholar] [CrossRef]
- Gobbo, F.; Fornasiero, D.; De Marco, M.A.; Zecchin, B.; Mulatti, P.; Delogu, M.; Terregino, C. Active Surveillance for Highly Pathogenic Avian Influenza Viruses in Wintering Waterbirds in Northeast Italy, 2020–2021. Microorganisms 2021, 9, 2188. [Google Scholar] [CrossRef]
- Pohlmann, A.; King, J.; Fusaro, A.; Zecchin, B.; Banyard, A.C.; Brown, I.H.; Byrne, A.M.P.; Beerens, N.; Liang, Y.; Heutink, R.; et al. Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021. mBio 2022, 13, e00609–e00622. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef]
- Tarasiuk, K.; Kycko, A.; Świętoń, E.; Bocian, Ł.; Wyrostek, K.; Śmietanka, K. Homo- and Heterosubtypic Immunity to Low Pathogenic Avian Influenza Virus Mitigates the Clinical Outcome of Infection with Highly Pathogenic Avian Influenza H5N8 Clade 2.3.4.4.b in Captive Mallards (Anas platyrhynchos). Pathogens 2023, 12, 217. [Google Scholar] [CrossRef]
- Animal and Plant Health Inspection Service; U.S. Department of Agriculture. 2022–2023 Detections of Highly Pathogenic Avian Influenza in Mammals. Available online: https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/animal-disease-information/avian/avian-influenza/hpai-2022/2022-hpai-mammals (accessed on 7 August 2023).
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Mirinavičiūtė, G.; Niqueux, É.; Staubach, C.; et al. Avian influenza overview June–September 2023. EFSA J. 2023, 21, e08328. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhu, W.; Yang, L.; Shu, Y. The Epidemiology, Virology, and Pathogenicity of Human Infections with Avian Influenza Viruses. Cold Spring Harb. Perspect. Med. 2021, 11, a038620. [Google Scholar] [CrossRef] [PubMed]
- Ellwanger, J.H.; Chies, J.A.B. Zoonotic spillover: Understanding basic aspects for better prevention. Genet. Mol. Biol. 2021, 44, e20200355. [Google Scholar] [CrossRef] [PubMed]
- Hoye, B.J.; Donato, C.M.; Lisovski, S.; Deng, Y.M.; Warner, S.; Hurt, A.C.; Klaassen, M.; Vijaykrishna, D. Reassortment and Persistence of Influenza A Viruses from Diverse Geographic Origins within Australian Wild Birds: Evidence from a Small, Isolated Population of Ruddy Turnstones. J. Virol. 2021, 95, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; Aznar, I.; Baldinelli, F.; Papanikolaou, A.; Stoicescu, A.; Van der Stede, Y. Annual Report on surveillance for avian influenza in poultry and wild birds in Member States of the European Union in 2020. EFSA J. 2021, 19, e06953. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; Aznar, I.; Baldinelli, F.; Stoicescu, A.; Kohnle, L. Annual report on surveillance for avian influenza in poultry and wild birds in Member States of the European Union in 2021. EFSA J. 2022, 20, e07554. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Regnaut, S.; Lucas, F.S.; Fumagalli, L. DNA degradation in avian faecal samples and feasibility of non-invasive genetic studies of threatened capercaillie populations. Conserv. Genet. 2006, 7, 449–453. [Google Scholar] [CrossRef]
- Vo, A.-T.E.; Jedlicka, J.A. Protocols for metagenomic DNA extraction and Illumina amplicon library preparation for faecal and swab samples. Mol. Ecol. Resour. 2014, 14, 1183–1197. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef] [PubMed]
- Panzarin, V.; Marciano, S.; Fortin, A.; Brian, I.; D’Amico, V.; Gobbo, F.; Bonfante, F.; Palumbo, E.; Sakoda, Y.; Le, K.T.; et al. Redesign and Validation of a Real-Time RT-PCR to Improve Surveillance for Avian Influenza Viruses of the H9 Subtype. Viruses 2022, 14, 1263. [Google Scholar] [CrossRef]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian real-time reverse transcriptase-polymerase chain reaction and its application in H5N1 outbreaks in 2005–2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Slomka, M.J.; Pavlidis, T.; Coward, V.J.; Voermans, J.; Koch, G.; Hanna, A.; Banks, J.; Brown, I.H. Validated RealTime reverse transcriptase PCR methods for the diagnosis and pathotyping of Eurasian H7 avian influenza viruses. Influenza Other Respir. Viruses 2009, 3, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Slomka, M.J.; Coward, V.J.; Banks, J.; Löndt, B.Z.; Brown, I.H.; Voermans, J.; Koch, G.; Handberg, K.J.; Jørgensen, P.H.; Cherbonnel-Pansart, M.; et al. Identification of sensitive and specific avian influenza polymerase chain reaction methods through blind ring trials organized in the European Union. Avian Dis. 2007, 51, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; Ahrens, A.K.; Ali, A.; El-Kady, M.F.; Hafez, H.M.; Mettenleiter, T.C.; Beer, M.; Harder, T. Improved Subtyping of Avian Influenza Viruses Using an RT-qPCR-Based Low Density Array: ‘Riems Influenza a Typing’, Version 2 (RITA-2). Viruses 2022, 14, 415. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Slomka, M.J.; Reid, S.M.; Thomas, S.S.; Mahmood, S.; Byrne, A.M.P.; Cooper, J.; Russell, C.; Mollett, B.C.; Agyeman-Dua, E.; et al. Development and Application of Real-Time PCR Assays for Specific Detection of Contemporary Avian Influenza Virus Subtypes N5, N6, N7, N8, and N9. Avian Dis. 2018, 63, 209–218, 210. [Google Scholar] [CrossRef] [PubMed]
- World Organisation for Animal Health. OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals. Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/tahm/3.03.04_AI.pdf (accessed on 17 October 2023).
- Istituto Zooprofilattico Sperimentale delle Venezie. Diagnostic Protocols. Available online: https://www.izsvenezie.com/reference-laboratories/avian-influenza-newcastle-disease/diagnostic-protocols/ (accessed on 18 October 2023).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic. Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, C.J.; Rajao, D.S.; Perez, D.R. Airborne Transmission of Avian Origin H9N2 Influenza A Viruses in Mammals. Viruses 2021, 13, 1919. [Google Scholar] [CrossRef]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.P.; Leung, Y.H.; Chow, C.K.; Ng, C.F.; Tsang, C.L.; Wu, Y.O.; Ma, S.K.; Sia, S.F.; Guan, Y.; Peiris, J.S. Identifying the species-origin of faecal droppings used for avian influenza virus surveillance in wild-birds. J. Clin. Virol. 2009, 46, 90–93. [Google Scholar] [CrossRef]
- Hebert, P.D.; Stoeckle, M.Y.; Zemlak, T.S.; Francis, C.M. Identification of Birds through DNA Barcodes. PLoS Biol. 2004, 2, e312. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D. bold: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, T.; Kawaoka, Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza A virus. J. Virol. 1994, 68, 3120–3128. [Google Scholar] [CrossRef]
- Senne, D.A.; Panigrahy, B.; Kawaoka, Y.; Pearson, J.E.; Süss, J.; Lipkind, M.; Kida, H.; Webster, R.G. Survey of the Hemagglutinin (HA) Cleavage Site Sequence of H5 and H7 Avian Influenza Viruses: Amino Acid Sequence at the HA Cleavage Site as a Marker of Pathogenicity Potential. Avian. Dis. 1996, 40, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Veits, J.; Weber, S.; Stech, O.; Breithaupt, A.; Gräber, M.; Gohrbandt, S.; Bogs, J.; Hundt, J.; Teifke, J.P.; Mettenleiter, T.C.; et al. Avian influenza virus hemagglutinins H2, H4, H8, and H14 support a highly pathogenic phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 2579–2584. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zu, Z.; Liu, J.; Song, J.; Wang, X.; Wang, C.; Liu, L.; Tong, Q.; Wang, M.; Sun, H.; et al. Prevailing I292V PB2 mutation in avian influenza H9N2 virus increases viral polymerase function and attenuates IFN-β induction in human cells. J. Gen. Virol. 2019, 100, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yuan, S.; Zhang, K.; Singh, K.; Ma, Q.; Zhou, J.; Chu, H.; Zheng, B.J. PB2 substitutions V598T/I increase the virulence of H7N9 influenza A virus in mammals. Virology 2017, 501, 92–101. [Google Scholar] [CrossRef]
- Elgendy, E.M.; Arai, Y.; Kawashita, N.; Daidoji, T.; Takagi, T.; Ibrahim, M.S.; Nakaya, T.; Watanabe, Y. Identification of polymerase gene mutations that affect viral replication in H5N1 influenza viruses isolated from pigeons. J. Gen. Virol. 2017, 98, 6–17. [Google Scholar] [CrossRef]
- Yamayoshi, S.; Yamada, S.; Fukuyama, S.; Murakami, S.; Zhao, D.; Uraki, R.; Watanabe, T.; Tomita, Y.; Macken, C.; Neumann, G.; et al. Virulence-affecting amino acid changes in the PA protein of H7N9 influenza A viruses. J. Virol. 2014, 88, 3127–3134. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, J.; Guo, J.; Deng, G.; Zhang, Q.; Wang, J.; He, X.; Wang, K.; Chen, J.; Li, Y.; et al. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. PLoS Pathog. 2014, 10, e1004508. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Ibrahim, M.S.; Ellakany, H.F.; Kawashita, N.; Mizuike, R.; Hiramatsu, H.; Sriwilaijaroen, N.; Takagi, T.; Suzuki, Y.; Ikuta, K. Acquisition of human-type receptor binding specificity by new H5N1 influenza virus sublineages during their emergence in birds in Egypt. PLoS Pathog. 2011, 7, e1002068. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, B.; Zhou, H.; Suguitan, A.L., Jr.; Cheng, X.; Subbarao, K.; Kemble, G.; Jin, H. Glycosylation at 158N of the hemagglutinin protein and receptor binding specificity synergistically affect the antigenicity and immunogenicity of a live attenuated H5N1 A/Vietnam/1203/2004 vaccine virus in ferrets. J. Virol. 2010, 84, 6570–6577. [Google Scholar] [CrossRef] [PubMed]
- Mancera Gracia, J.C.; Van den Hoecke, S.; Richt, J.A.; Ma, W.; Saelens, X.; Van Reeth, K. A reassortant H9N2 influenza virus containing 2009 pandemic H1N1 internal-protein genes acquired enhanced pig-to-pig transmission after serial passages in swine. Sci. Rep. 2017, 7, 1323. [Google Scholar] [CrossRef] [PubMed]
- Kimble, J.B.; Sorrell, E.; Shao, H.; Martin, P.L.; Perez, D.R. Compatibility of H9N2 avian influenza surface genes and 2009 pandemic H1N1 internal genes for transmission in the ferret model. Proc. Natl. Acad. Sci. USA 2011, 108, 12084–12088. [Google Scholar] [CrossRef]
- Kode, S.S.; Pawar, S.D.; Tare, D.S.; Keng, S.S.; Hurt, A.C.; Mullick, J. A novel I117T substitution in neuraminidase of highly pathogenic avian influenza H5N1 virus conferring reduced susceptibility to oseltamivir and zanamivir. Vet. Microbiol. 2019, 235, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef]
- European Food Safety Authority; European Centre for Disease Prevention Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Avian influenza overview September–December 2021. EFSA J. 2021, 19, e07108. [Google Scholar] [CrossRef]
- Istituto Zooprofilattico Sperimentale delle Venezie. Highly Pathogenic Avian Influenza in birds other than poultry in Italy. Epidemiological Situation 1 March 2022. Available online: https://www.izsvenezie.com/documents/reference-laboratories/avian-influenza/italy-updates/HPAI/2021-1/italy-wildbirds.pdf (accessed on 22 October 2023).
- Verhagen, J.H.; Fouchier, R.A.M.; Lewis, N. Highly Pathogenic Avian Influenza Viruses at the Wild-Domestic Bird Interface in Europe: Future Directions for Research and Surveillance. Viruses 2021, 13, 212. [Google Scholar] [CrossRef] [PubMed]
- Hood, G.; Roche, X.; Brioudes, A.; von Dobschuetz, S.; Fasina, F.O.; Kalpravidh, W.; Makonnen, Y.; Lubroth, J.; Sims, L. A literature review of the use of environmental sampling in the surveillance of avian influenza viruses. Transbound Emerg. Dis. 2021, 68, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Nazir, J.; Haumacher, R.; Ike, A.C.; Marschang, R.E. Persistence of avian influenza viruses in lake sediment, duck feces, and duck meat. Appl. Environ. Microbiol. 2011, 77, 4981–4985. [Google Scholar] [CrossRef] [PubMed]
- Animal and Plant Health Inspection Service; U.S. Department of Agriculture. 2022–2023 Detections of Highly Pathogenic Avian Influenza in Wild Birds. Available online: https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/animal-disease-information/avian/avian-influenza/hpai-2022/2022-hpai-wild-birds (accessed on 22 October 2023).
- Graziosi, G.; Lupini, C.; Dalla Favera, F.; Martini, G.; Dosa, G.; Garavini, G.; Trevisani, G.; Mannelli, A.; Catelli, E. Characterization and quantification of wildlife visits to commercial poultry farms, assessed by camera traps in an area at high risk of introduction of avian influenza. In Proceedings of the XXIInd Congress of the World Veterinary Poultry Association (WVPAC), Verona, Italy, 4–8 September 2023; p. 171. [Google Scholar]
- Graziosi, G.; Lupini, C.; Gobbo, F.; Zecchin, B.; Garavini, G.; Trevisani, G.; Dosa, G.; Mannelli, A.; Terregino, C.; Catelli, E. Characterizing the interface between wild birds and poultry in a high-risk area for avian influenza introduction: Camera traps in poultry farms and virological surveillance in waterfowl. In Proceedings of the American Association of Avian Pathologists (AAAP) 2023 Annual Meeting, Jacksonville, FL, USA, 11–14 June 2023. [Google Scholar]
- Ahrens, A.K.; Selinka, H.C.; Wylezich, C.; Wonnemann, H.; Sindt, O.; Hellmer, H.H.; Pfaff, F.; Höper, D.; Mettenleiter, T.C.; Beer, M.; et al. Investigating Environmental Matrices for Use in Avian Influenza Virus Surveillance-Surface Water, Sediments, and Avian Fecal Samples. Microbiol. Spectr. 2023, 11, e0266422. [Google Scholar] [CrossRef]
- Pawar, S.D.; Pande, S.A.; Tare, D.S.; Keng, S.S.; Kode, S.S.; Singh, D.K.; Mullick, J. Morphological and Biochemical Characteristics of Avian Faecal Droppings and Their Impact on Survival of Avian Influenza Virus. Food Environ. Virol. 2018, 10, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Busquets, N.; Alba, A.; Napp, S.; Sánchez, A.; Serrano, E.; Rivas, R.; Núñez, J.I.; Majó, N. Influenza A virus subtypes in wild birds in North-Eastern Spain (Catalonia). Virus Res. 2010, 149, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.M.; Fagulha, T.; Barros, S.C.; Ramos, F.; Duarte, M.; Luís, T.; Fevereiro, M. Multiyear surveillance of influenza A virus in wild birds in Portugal. Avian. Pathol. 2011, 40, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Lebarbenchon, C.; Albespy, F.; Brochet, A.L.; Grandhomme, V.; Renaud, F.; Fritz, H.; Green, A.J.; Thomas, F.; van der Werf, S.; Aubry, P.; et al. Spread of avian influenza viruses by common teal (Anas crecca) in Europe. PLoS ONE 2009, 4, e7289. [Google Scholar] [CrossRef] [PubMed]
- Lebarbenchon, C.; van der Werf, S.; Thomas, F.; Aubin, J.T.; Azebi, S.; Cuvelier, F.; Jeannin, P.; Roca, V.; Chang, C.M.; Kayser, Y.; et al. Absence of detection of highly pathogenic H5N1 in migratory waterfowl in southern France in 2005–2006. Infect. Genet. Evol. 2007, 7, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; Baas, C.; Lexmond, P.; Waldenström, J.; Wallensten, A.; Fransson, T.; Rimmelzwaan, G.F.; Beyer, W.E.P.; Schutten, M.; Olsen, B.; et al. Spatial, Temporal, and Species Variation in Prevalence of Influenza A Viruses in Wild Migratory Birds. PLoS Pathog. 2007, 3, e61. [Google Scholar] [CrossRef]
- Tanikawa, T.; Sakuma, S.; Yoshida, E.; Tsunekuni, R.; Nakayama, M.; Kobayashi, S. Comparative susceptibility of the common teal (Anas crecca) to infection with high pathogenic avian influenza virus strains isolated in Japan in 2004–2017. Vet. Microbiol. 2021, 263, 109266. [Google Scholar] [CrossRef] [PubMed]
- van den Brand, J.M.A.; Verhagen, J.H.; Veldhuis Kroeze, E.J.B.; van de Bildt, M.W.G.; Bodewes, R.; Herfst, S.; Richard, M.; Lexmond, P.; Bestebroer, T.M.; Fouchier, R.A.M.; et al. Wild ducks excrete highly pathogenic avian influenza virus H5N8 (2014–2015) without clinical or pathological evidence of disease. Emerg. Microbes Infect. 2018, 7, 67. [Google Scholar] [CrossRef] [PubMed]
- Barkhasbaatar, A.; Gilbert, M.; Fine, A.E.; Shiilegdamba, E.; Damdinjav, B.; Buuveibaatar, B.; Khishgee, B.; Johnson, C.K.; Leung, C.Y.H.; Ankhanbaatar, U.; et al. Ecological characterization of 175 low-pathogenicity avian influenza viruses isolated from wild birds in Mongolia, 2009–2013 and 2016–2018. Vet. Med. Sci. 2023, 9, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Torrontegi, O.; Alvarez, V.; Acevedo, P.; Gerrikagoitia, X.; Höfle, U.; Barral, M. Long-term avian influenza virus epidemiology in a small Spanish wetland ecosystem is driven by the breeding Anseriformes community. Vet. Res. 2019, 50, 4. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Ma, E.J.; Meixell, B.W.; Lindberg, M.S.; Boyce, W.M.; Runstadler, J.A. Transmission of influenza reflects seasonality of wild birds across the annual cycle. Ecol. Lett. 2016, 19, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Latorre-Margalef, N.; Grosbois, V.; Wahlgren, J.; Munster, V.J.; Tolf, C.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Olsen, B.; Waldenström, J. Heterosubtypic Immunity to Influenza A Virus Infections in Mallards May Explain Existence of Multiple Virus Subtypes. PLoS Pathog. 2013, 9, e1003443. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, J.G.; Hoye, B.J.; Verhagen, J.H.; Nolet, B.A.; Fouchier, R.A.; Klaassen, M. Juveniles and migrants as drivers for seasonal epizootics of avian influenza virus. J. Anim. Ecol. 2014, 83, 266–275. [Google Scholar] [CrossRef]
- Latorre-Margalef, N.; Brown, J.D.; Fojtik, A.; Poulson, R.L.; Carter, D.; Franca, M.; Stallknecht, D.E. Competition between influenza A virus subtypes through heterosubtypic immunity modulates re-infection and antibody dynamics in the mallard duck. PLoS Pathog. 2017, 13, e1006419. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef]
- Briand, F.X.; Schmitz, A.; Ogor, K.; Le Prioux, A.; Guillou-Cloarec, C.; Guillemoto, C.; Allée, C.; Le Bras, M.O.; Hirchaud, E.; Quenault, H.; et al. Emerging highly pathogenic H5 avian influenza viruses in France during winter 2015/16: Phylogenetic analyses and markers for zoonotic potential. Euro. Surveill. 2017, 22, 30473. [Google Scholar] [CrossRef]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, E.; Staubach, C.; et al. Avian influenza overview March–June 2022. EFSA J. 2022, 20, e07415. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority; European Centre for Disease Prevention and Control; European Union Reference Laboratory for Avian Influenza; Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Mirinaviciute, G.; Niqueux, E.; et al. Avian influenza overview December 2022–March 2023. EFSA J. 2023, 21, e07917. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Mettenleiter, T.C.; Abdelwhab, E.M. A brief summary of the epidemiology and genetic relatedness of avian influenza H9N2 virus in birds and mammals in the Middle East and North Africa. Epidemiol. Infect. 2017, 145, 3320–3333. [Google Scholar] [CrossRef] [PubMed]
- Carnaccini, S.; Perez, D.R. H9 Influenza Viruses: An Emerging Challenge. Cold Spring Harb. Perspect Med. 2020, 10, a038588. [Google Scholar] [CrossRef] [PubMed]
- Sagong, M.; Lee, K.N.; Lee, E.K.; Kang, H.; Choi, Y.K.; Lee, Y.J. Current situation and control strategies of H9N2 avian influenza in South Korea. J. Vet. Sci. 2023, 24, e5. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Krauss, S.; Webster, R.G. H9N2 Influenza A Viruses from Poultry in Asia Have Human Virus-like Receptor Specificity. Virology 2001, 281, 156–162. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AIV Positive/Sample Size (Positivity Rate %) | AIV Subtype | |
|---|---|---|
| Bird migration phenology period | ||
| Autumn migration † | 2/203 (1) | HxN9, H5N3 |
| Wintering †† | 2/134 (1.5) | H9N2, HxN2 |
| Spring migration ††† | 0/70 (0) | n.a. ° |
| Bird taxa of origin | ||
| Ducks | 2/249 (0.8) | HxN9, H5N3 |
| Geese | 1/46 (2.1) | H9N2 |
| Ibises | 0/18 (0) | n.a. |
| Gulls | 0/19 (0) | n.a. |
| Herons | 0/30 (0) | n.a. |
| Flamingos | 0/1 (0) | n.a. |
| Shorebirds | 1/34 (3.0) | HxN2 |
| Rails | 0/10 (0) | n.a. |
| Total | 4/407 (1.0) | HxN9, H5N3, H9N2, HxN2 |
| Bird Species | AIV Positive/Sample Size (Positivity Rate %) | AIV Subtype |
|---|---|---|
| Anseriformes order | ||
| Eurasian teal (Anas crecca) | 7/85 (8.2) | H1N1, HxN9, H9N2, H6N1 |
| Mallard (Anas platyrhynchos) | 0/8 (0) | n.a. ° |
| Greylag goose (Anser anser) † | 0/3 (0) | n.a. |
| Eurasian wigeon (Mareca penelope) | 0/1 (0) | n.a. |
| Gadwall (Mareca strepera) | 0/2 (0) | n.a. |
| Northern shoveler (Spatula clypeata) | 0/22 (0) | n.a. |
| Charadriiformes order | ||
| Common snipe (Gallinago gallinago) | 0/2 (0) | n.a. |
| Spotted redshank (Tringa erythropus) † | 0/1 (0) | n.a. |
| Northern lapwing (Vanellus vanellus) † | 0/5 (0) | n.a. |
| Total | 7/129 (5.4) | H1N1, HxN9, H9N2, H6N1 |
| Viral Protein | Amino Acid Substitution | A/Teal/Italy/1821-10_22VIR4622-1/2021 (H1N1) | A/Mallard/Italy/22VIR4203-2/2021 (H5N3) | A/Teal/Italy/1856-7_22VIR4622-7/2021 (H9N2) | A/Teal/Italy/1828-6_22VIR4622-5/2021 (H9N2) | A/Teal/Italy/1821-14_22VIR4622-3/2021 (H9N2) | Phenotype (Subtype Tested) | Ref. |
|---|---|---|---|---|---|---|---|---|
| PB2 | I292V a | I | I | V | n.a. * | n.a. * | Increased polymerase activity in mammalian cell line, increased virulence in mice (H9N2) | [53] |
| V598T/I | T | T | T | T | T | Increased polymerase activity and replication in mammalian cells, increased virulence in mice (H7N9) | [54] | |
| PB1 | D3V a | V | V | V | V | V | Increased polymerase activity and viral replication in avian and mammalian cell lines (H5N1) | [55] |
| PA | S37A a | A | A | A | A | A | Increased polymerase activity in mammalian cell line (H7N9) | [56] |
| HA | I155T b | I | T | T | T | T | Increased binding to mammal-like receptor (H5N1, H9N2) | [57,58] |
| S159N | N | N | N | N | N | Increased binding to mammal-like receptor (H5N1) | [59] | |
| D225G | G | G | G | G | G | Increased transmission and replication in swine (H1N1 backbone with HA and NA of H9N2) | [60] | |
| NA | A30T c | I | I | T | T | T | Observed in airborne transmission in ferrets (H9N2) | [61] |
| I117T | I | T | T | T | T | Reduced susceptibility to oseltamivir and zanamivir (H5N1) | [62] | |
| NS1 | P42S a | S | S | S | S | S | Increased virulence and decreased antiviral response in mice (H5N1) | [63] |
| M1 | N30D a | D | D | D | D | D | Increased virulence in mice (H5N1) | [64] |
| AIV Strain | Gene Segment | Highest Homology AIV Strain | Accession Number | % Homology |
|---|---|---|---|---|
| A/teal/Italy/1821-10_22VIR4622-1/2021 (H1N1) | PB2 | A/Anas_platyrhynchos/Belgium/1098_2/2020(H1N1) | EPI1942951 | 99 |
| PB1 | A/Anas_platyrhynchos/Belgium/2213_0006/2021(H11N6) | EPI2122874 | 99 | |
| PA | A/Anas_platyrhynchos/Belgium/1098_2/2020(H1N1) | EPI1942953 | 98 | |
| HA | A/Anas_platyrhynchos/Belgium/1098_2/2020(H1N1) | EPI1942954 | 99 | |
| NP | A/Anas platyrhynchos/Belgium/11025_44/2017 (H11N1) | EPI1774315 | 99 | |
| NA | A/chicken/Denmark/S02750-3/2020(H5N1) | EPI1694135 | 98 | |
| M | A/duck/Moscow/5881/2021(H3N2) | EPI2175875 | 99 | |
| NS | A/Environment/Jiangxi/12590/2019 (H10N3) | EPI1848446 | 99 | |
| A/mallard/Italy/22VIR4203-2/2021 (H5N3) | PB2 | A/teal/Italy/21VIR8923-73/2021(H5N3) | EPI7987343 | 99 |
| PB1 | A/duck/Italy/21VIR8022-1/2021(H5N3) | EPI1946718 | 99 | |
| PA | A/duck/Italy/21VIR8022-1/2021(H5N3) | EPI1946719 | 99 | |
| HA | A/duck/Italy/21VIR8022-1/2021(H5N3) | EPI1946720 | 99 | |
| NP | A/duck/Italy/21VIR8022-1/2021(H5N3) | EPI1946721 | 99 | |
| NA | A/duck/Italy/21VIR8022-1/2021(H5N3) | EPI1946726 | 99 | |
| M | A/teal/Italy/21VIR8923-73/2021(H5N3) | EPI1947344 | 99 | |
| NS | A/duck/Italy/21VIR8022-1/2021 (H5N3) | EPI1946723 | 99 | |
| A/teal/Italy/1856-7_22VIR4622-7/2021 (H9N2); A/teal/Italy/1828-6_22VIR4622-5/2021 (H9N2); A/teal/Italy/1821-14_22VIR4622-3/2021 (H9N2). | PB2 | A/Anas platyrhynchos/Belgium/10402_H195386/2017 (H1N1) | EPI1775505 | 98 |
| PB1 | A/duck/Italy/21VIR8024-4/2021(H5N3) | EPI1947293 | 99 | |
| PA | A/shoveler/Novosibirsk region/3465k/2020 (H3N8) | EPI1849977 | 98 | |
| HA | A/mallard/Poland/P096/2020(H9N2) | EPI2618292 | 98 | |
| NP | A/Mallard/Netherlands/37/2015(H3N8) | EPI1530590 | 98 | |
| NA | A/environment/England/030642/2020 (H5N2) | EPI2062134 | 98 | |
| M | A/pintail/Egypt/MB-D-384C/2015(H3N6) | EPI1581277 | 98 | |
| NS | A/mallard duck/Netherlands/41/2015 (H5N1) | EPI1306985 | 98 |
| Gene | AIV Strain | Subtype | Amino Acid Substitutions * | Source | |||
|---|---|---|---|---|---|---|---|
| This Study | Best Similar | This Study | Best Similar | This Study | Best Similar | ||
| HA | A/teal/Italy/1821-10_22VIR4622-1/2021 | A/Anas_platyrhynchos/ Belgium/1098_2/2020 | H1N1 | H1N1 | 456 (W → R) | Eurasian teal, Bologna province, 15 October 2021 | Mallard, Belgium, 12 September 2020 |
| NA | A/teal/Italy/ 1821-10_22VIR4622-1/2021 | A/chicken/Denmark/ S02750-3/2020 | H1N1 | H5N1 (LPAIV) | 79 (I → V) 83 (T → A) 283 (M → I) | Eurasian teal, Bologna province, 15 October 2021 | Chicken, Denmark, 28 January 2020 |
| HA | A/mallard/Italy/ 22VIR4203-2/2021 | A/duck/Italy/ 21VIR8022-1/2021 | H5N3 (LPAIV) | H5N3 (LPAIV) | 81 (K → N) 395 (I → V) | Mallard, Emilia-Romagna region, 13 October 2021 | Duck, Italy, Lombardy region, 23 September 2021 |
| NA | A/mallard/Italy/ 22VIR4203-2/2021 | A/duck/Italy/ 21VIR8022-1/2021 | H5N3 (LPAIV) | H5N3 (LPAIV) | 169 (V → I) | Mallard, Emilia-Romagna region, 13 October 2021 | Duck; Italy, Lombardy region; 23 September 2021 |
| HA | A/teal/Italy/1856-7_22VIR4622-7/2021(H9N2) ° | A/mallard/Poland/ P096/2020 | H9N2 | H9N2 | 245 (I → V) 419 (D → N) | Eurasian teal; Bologna province, October–November 2021 | Duck; Poland; 2 of November 2020 |
| NA | A/teal/Italy/1856-7_22VIR4622-7/2021(H9N2) ° | A/environment/England/ 030642/2020 | H9N2 | H5N2 (LPAIV) | 43 (S → N) 216 (G → V) 262 (V → I) 302 (I → V) 313 (D → G) | Eurasian teal, Bologna province, October–November 2021 | Faeces, England, 31 of October 2020 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graziosi, G.; Lupini, C.; Gobbo, F.; Zecchin, B.; Quaglia, G.; Pedrazzoli, S.; Lizzi, G.; Dosa, G.; Martini, G.; Terregino, C.; et al. Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies. Animals 2024, 14, 1018. https://doi.org/10.3390/ani14071018
Graziosi G, Lupini C, Gobbo F, Zecchin B, Quaglia G, Pedrazzoli S, Lizzi G, Dosa G, Martini G, Terregino C, et al. Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies. Animals. 2024; 14(7):1018. https://doi.org/10.3390/ani14071018
Chicago/Turabian StyleGraziosi, Giulia, Caterina Lupini, Federica Gobbo, Bianca Zecchin, Giulia Quaglia, Sara Pedrazzoli, Gabriele Lizzi, Geremia Dosa, Gabriella Martini, Calogero Terregino, and et al. 2024. "Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies" Animals 14, no. 7: 1018. https://doi.org/10.3390/ani14071018
APA StyleGraziosi, G., Lupini, C., Gobbo, F., Zecchin, B., Quaglia, G., Pedrazzoli, S., Lizzi, G., Dosa, G., Martini, G., Terregino, C., & Catelli, E. (2024). Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies. Animals, 14(7), 1018. https://doi.org/10.3390/ani14071018