Simple Summary
As an endemic and vulnerable fish from the upper Yangtze River in China, Schizothorax kozlovi holds significant scientific and ecological importance, yet it has received little attention so far. In this paper, we reported the characterization of the mitochondrial genome of S. kozlovi, and further investigated the phylogenetic relationships of Schizothorax. The results showed that the mitochondrial genome of S. kozlovi had a total size of 16,585 bp, a circular arrangement, and contained 13 PCGs, 22 tRNAs, two rRNAs, and two non-coding regions. Moreover, the phylogenetic analyses demonstrated that Schizothorax could be classified into four clades, and S. kozlovi was closely related to Schizothorax chongi. The present study enriched the basic biological data for S. kozlovi and provided fundamental references for the conservation of S. kozlovi and Schizothorax.
Abstract
Schizothorax kozlovi is an endemic and vulnerable fish species found in the upper Yangtze River in China. Over the past few years, the population resources of S. kozlovi have been nearly completely depleted owing to multiple contributing threats. While the complete mitochondrial genomes serve as important molecular markers for phylogenetic and genetic studies, the mitochondrial genome of S. kozlovi has still received little attention. In this study, we analyzed the characterization of the mitochondrial genome of S. kozlovi and investigated the phylogenetic relationships of Schizothorax. The complete mitochondrial genome of S. kozlovi was 16,585 bp in length, which contained thirty-seven genes (thirteen protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), twenty-two transfer RNA genes (tRNAs)) and two non-coding regions for the origin of light strand (OL) and the control region (CR). There were nine overlapping regions and seventeen intergenic spacers regions in the mitochondrial genome. The genome also showed a bias towards A + T content (55.01%) and had a positive AT-skew (0.08) and a negative GC-skew (−0.20). All the PCGs employed the ATG or GTG as the start codon and TAA, TAG, or single T as the stop codon. Additionally, all of the tRNAs displayed a typical cloverleaf secondary structure, except trnS1 which lacked the D arm. The phylogenetic analysis, based on the maximum likelihood (ML) and Bayesian inference (BI) methods, revealed that the topologies of the phylogenetic tree divided the Schizothorax into four clades and did not support the classification of Schizothorax based on morphology. The phylogenetic status of S. kozlovi was closely related to that of S. chongi. The present study provides valuable genomic information for S. kozlovi and new insights in phylogenetic relationships of Schizothorax. These data could also offer fundamental references and guidelines for the management and conservation of S. kozlovi and other species of Schizothorax.
1. Introduction
The mitochondria, as fundamental organelles, are widely found in eukaryotic cells and are associated with pivotal roles such as energy metabolism, genetic signaling, biological synthesis, and cell apoptosis [1]. Since the discovery of the mitochondria, researchers have never ceased to explore their structure and function. Particularly with the advancement of molecular biotechnology in recent years, the mitochondrial genome has received a great deal of attention. Several studies have shown that the mitochondria of animals contain an independent genome and that the mitochondrial genome is a closed-circular DNA molecule ranging in size from 14 kb to 20 kb with a high degree of autonomy in terms of genetic replication, transmission, and expression [2]. The mitochondrial genome typically consists of thirty-seven genes, including thirteen protein-coding genes (PCGs), twenty-two transfer RNA genes (tRNAs), two ribosomal RNA genes (12S rRNA and 16S rRNA), and two non-coding regions (origin of the light strand (OL) and the control region (CR)), except for a few species (e.g., Crassostrea gigas, Metridium senile) with variations in the characterization of the genome [3]. Moreover, mitochondrial genome sequencing analysis revealed that the mitochondrial DNA (mtDNA) is a double-stranded molecule with the heavy chain on the outer ring and the light chain on the inner ring, but the majority of genes are transcribed by the heavy chain. The rapid advances in sequencing technology over the past few years have facilitated the availability of the mtDNA, which has led to a boom in the field of animal mitochondrial genome research, particularly in vertebrates such as fishes, amphibians, reptiles, and birds [4,5,6,7]. As the study progressed, researchers have summarized that mtDNA is characterized by small size, simple structure, maternal inheritance, missing intron, high evolutionary rate, conserved content, and easy amplification [8,9,10]. Based on the advantages of genomic characterization above, the mtDNA has been employed as a useful molecular marker in the study of molecular evolution, biodiversity analysis, phylogenetic reconstruction, population genetic assessment, and genomic comparison [11,12]. In the past decade, there also has been a flourish in the sequencing of fish mitochondrial genomes. For example, analyses of gene localizations, tRNA secondary structures, and the transcription and replication regions within the mitochondrial genome have provided a wealth of reliable information on fish taxonomic classification, ecological adaptation, and phylogenetic relationships, reminding us that the complete mitochondrial genome are important molecular resources for the management and conservation of fishes [13].
Schizothorax kozlovi Nikolsky, 1903 belongs to Cypriniformes, Cyprinidae, Schizothorax and is mainly distributed in the Jinsha River, Yalong River, and Wujiang River basins [14]. In addition, S. kozlovi is considered to be a typical plateau cold-water fish and it is also an endemic and economic fish found in the upper Yangtze River in China. The S. kozlovi has been an important fish with great value in ecology, the economy, and scientific research. Unfortunately, recent field surveys have revealed that population resources of S. kozlovi were significantly decreased and close to being depleted due to overfishing, water pollution, climate change, and habitat fragmentation [15]. S. kozlovi has also been recognized as a vulnerable species (VU) in the IUCN Red List [16]. Therefore, research on and attention to the biology of S. kozlovi are urgently needed for sustainable resource conservation. Previously, researchers have conducted studies on growth, reproduction, diet, population, and diversity [15,17,18,19,20,21]. But there has been little focus on the characterization of the mitochondrial genome and phylogenetic analysis for S. kozlovi. Only a few sequences of mitochondrial genome are included in the GenBank database (e.g., NC 027670.1) and Wang et al. [22] incompletely explored the phylogenetic relationships of some fish species in the Schizothorax. Morphologically, the S. kozlovi had some unique features such as a long and flat body covered in tiny scales, an inconspicuous keratin at the anterior edge of the jaw, a strong dorsal fin, two pairs of beards, and several pairs of developed anal scales [14,23]. These traits provided the basis for behaviors associated with adaptation to the low temperature and hypoxia of the plateau habitats [24,25]. In addition, the mitochondrial genome which encodes its functional genes might provide some key evidence about its adaptability, yet the complete mitochondrial genome data of S. kozlovi have rarely been reported and analyzed.
Over the past century, the fish species Schizothorax has received great interest from researchers. The Schizothorax is widely distributed across the Tibetan Plateau and is a representative example of the adaptation of native fish species to the plateau habitat. Several studies have suggested that the evolution process of Schizothorax is closely related to the uplift process in the Tibetan Plateau [26,27,28]. Specifically, Chen and Cao [29] summarized that the Schizothorax is the largest genus for the subfamily of Schizothoracinae living in China. Hence, Schizothorax is an excellent model to explore the process of fish species’ evolution and differentiation in a complex habitat. However, the controversies over the taxonomic and phylogenetic relationships of the Schizothorax have long existed because of their extreme morphological and ecological similarity. For example, Heckel [30] recommend that the Schizothorax can be divided into three groups based on the shape of the head and the morphology of the mouth; Chen et al. [31] suggested that the Schizothorax should be classified into two subgenera on the basis of the presence or absence of keratin at the anterior edge of the jaw. Moreover, researchers have previously used single mitochondrial gene such as Cyt b or COI to determine phylogenetic relationships for species among Schizothorax, which may lead to one-sided and biased conclusions [21,22,32]. Within this context, it is necessary to unite multiple makers from genomes to investigate the taxonomic and phylogenetic relationships for Schizothorax.
In this study, we reported the complete mitochondrial genome sequence of S. kozlovi; described the characterization of mitochondrial genome such as genome composition, organization, annotation; and reconstructed a phylogenetic tree based on the sequences within the mitochondrial genome. From this, we aimed to comprehensively understand the complete mitochondrial genome of S. kozlovi, to determine the potential phylogenetic status of S. kozlovi, and to explore the phylogenetic relationships of Schizothorax. Therefore, the results will provide fundamental molecular data for resources management and the conservation of S. kozlovi and help us form a better understanding of the phylogenetic relationships of Schizothorax.
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
The specimen of S. kozlovi was collected from the Zangqu River (31°34′ N, 98°24′ E), a tributary of the upper Jinsha River, China. Once captured, the specimen was identified to species level according to the main ideas about morphology referring to Ding [14], Chen [31], and Wu [23]. Then, 2–3 g of muscle tissue was cut from the left side of the back and tagged with a label for the specimen. Subsequently, the specimen and muscle tissue were immediately preserved in 95% ethanol and kept in a refrigerator at −20 °C until taken back to the laboratory. Finally, the specimen and muscle tissue were stored in the College of Life Science at China West Normal University until DNA extraction. The experiments on S. kozlovi in this study were conducted in accordance with the national guidelines for laboratory animal care and treatment. This study was approved by the Institutional Review Board of China West Normal University.
The total genomic DNA from left back muscle tissue was extracted by using E.Z.N.A.® Tissue DNA Kit (Origin gene Co. Ltd., Shanghai, China) in accordance with the manufacturer’s guidelines. The genomic DNA was analyzed with 1% agarose gel electrophoresis to detect the DNA quality. And the extracted DNA was stored at −20 °C for subsequent use.
2.2. Genome Sequencing and Assembly
In this study, next-generation sequencing (NGS) was employed to obtain the complete mitochondrial genome sequence of S. kozlovi. Firstly, the genomic DNA was fragmented into 300–500 bp size fragments by Covaris M220, target fragments were amplified by polymerase chain reaction (PCR), and the library was constructed by TruseqTM RNA sample Prep Kit (Illumina, San Diego, CA, USA) with 1 µg DNA. Then, the mitochondrial genome sequence was performed on Illumina NovaSeq6000 platform at Origin gene Co. Ltd., Shanghai, China. Raw reads were converted from Illumina sequencing into FASTQ format by FastQC v0.11.4 [33]. Moreover, the raw data were cleaned up so that the low quality reads (<Q20), high “N” ratio sequences (>10%), fragments with length less than 50 bp were filtered out to obtain high quality clean data for quality control [34]. And multiple iterations of splicing for clean data were performed by Getorganelle v1.7.0 [35] and corrected by using Pilon v1.23 [36]. Finally, the clean data were assembled with the de novo assembly program to obtain complete mitochondrial genome sequence by NOVOPlasty v4.2 [37].
2.3. Sequence Annotation and Analysis
The mitochondrial genome of PCGs, rRNAs, and tRNAs were annotated in the MITOS2 webserver (http://mitos2.bioinf.uni-leipzig.de/index.py, accessed on 23 December 2021) [38]. The secondary structures of tRNAs from S. kozlovi were initially predicted by the tRNAscan webserver (http://lowelab.ucsc.edu/tRNAscan-SE/, accessed on 18 May 2023) [39]. The circular mitochondrial genome map was generated by OGDRAW v1.2 [40]. The relative synonymous codon usage (RSCU) for protein-coding genes, start/stop codon, and nucleotide composition were analyzed in the MEGA v7.0 [41] and the Codonw v1.4.4 (https://sourceforge.net/projects/codonw/, accessed on 23 December 2021). The skew values of the nucleotide composition was measured based on the following formulas: AT-skew = (A − T)/(A + T) and GC-skew = (G − C)/(G + C), where a positive AT-skew/GC-skew value represents that there are more As than Ts; while a negative AT-skew/GC-skew value means that there are less As than Ts [42,43]. The complete mitochondrial genome sequence of S. kozlovi was submitted to the GenBank database with accession number OR416862.1.
2.4. Phylogenetic Analysis
To clarify the phylogenetic status of S. kozlovi and understand the phylogenetic relationships within Schizothorax, available mitochondrial genome sequences for 32 species of Schizothorax and two species of Spinibarbus denticulatus and Spinibarbus sinensis from Spinibarbus as outgroup were downloaded from the GenBank database (https://www.ncbi.nlm.nih.gov/, accessed on 10 January 2024) (Table 1). The nucleotide sequences of thirteen PCGs, twenty-two tRNAs, and two rRNAs from mitochondrial genome for all species (a total of 35 species including S. kozlovi) were employed to reconstruct the phylogenetic tree. In this study, all procedures, including sequence extraction, alignment, and concatenation were performed in Phylosuite v1.2.1, which integrated multiple software programs for phylogenetic analyses [44]. After that, based on the concatenated sequence matrix, Bayesian inference (BI) and maximum likelihood (ML) methods were applied to determine phylogenetic trees. The selection of the best-fit substitution model for phylogenetic analyses was carried out by ModelFinder integrated in Phylosuite [45]. The maximum likelihood (ML) analysis was inferred by IQ-TREE integrated in Phylosuite, which applied the automatically selected option of the model in IQ-TREE for 5000 ultrafast bootstrap replicates [46,47]. The Bayesian inference (BI) analysis was deduced by MrBayes integrated in Phylosuite with four simultaneous Markov chain Monte Carlo (MCMC) algorithms [48]. Two independent runs of 1,000,000 generations were performed for sampling every 1000 generations, in which the initial 25% data were discarded as burn in and BI analysis was considered to be achieved since the average standard deviation of split frequencies was below 0.01 [49]. The final phylogenetic trees were viewed in iTOL v6.8.1 (https://itol.embl.de/, accessed on 2 February 2024).

Table 1.
The list of species included in this study for phylogenetic analyses.
3. Results
3.1. Genome Organization and Composition
The complete mitochondrial genome of S. kozlovi is a closed, circular molecule with a total length of 16,585 bp (Figure 1). And the obtained mitochondrial genome contained thirty-seven genes, including thirteen protein-coding genes (PCGs), two ribosomal RNA genes (rRNAs), twenty-two transfer RNA genes (tRNAs), and two non-coding regions for the origin of light strand (OL) and control region (CR) (Table 2). Among these genes, nine genes with the one protein-coding gene nad6 and eight transfer RNA genes, trnQ, trnA, trnN, trnC, trnY, trnS2, trnE, trnP, were encoded on the light strand (L), while twenty-eight genes including twelve protein-coding genes, fourteen transfer RNA genes, and two ribosomal RNA genes were located on the heavy strand (H) (Figure 1). Specifically, there were nine overlapping regions in the mitochondrial genome of S. kozlovi, which comprised a total length of 29 bp and varied in size from 1 to 7 bp, and the longest overlapping regions were located between the atp8 and atp6 and nad4l and nad4 genes (Table 2). In addition, 17 intergenic spacers regions with a total length of 201 bp were found in the mitochondrial genome, which ranged in size from 1 to 102 bp and the largest intergenic spacer region lied between CR and trnF genes (Table 2).
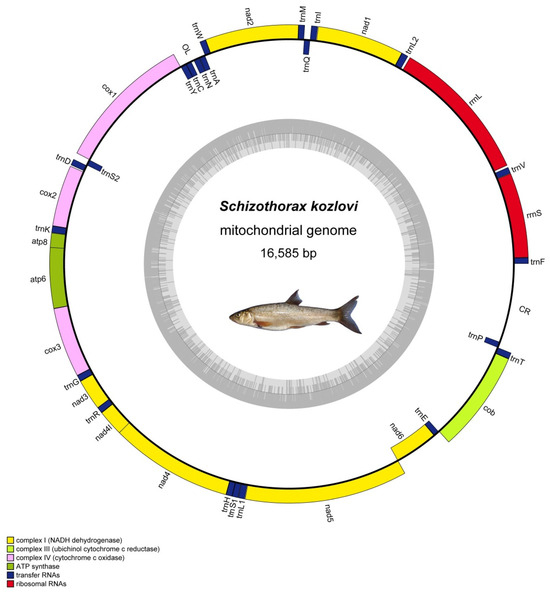
Figure 1.
A circular map of the mitochondrial genome of S. kozlovi. The inner ring is light strand and the outer ring is heavy strand. The thirteen PCGs, two rRNAs, twenty-two tRNAs, and two non-coding regions of OL and CR are shown on the circular map of mitochondrial genome. The blue strips represent transfer RNA genes; the red strips represent ribosomal RNA genes; the yellow strips, light green strips, pink strips, dark green strips represent protein-coding genes for NADH dehydrogenase, cytochrome c oxidase, cytochrome b, ATP synthase, respectively.
Table 2.
The organization of the complete mitochondrial genome of S. kozlovi. The thirteen PCGs (nad1, nad2, cox1, cox2, atp8, atp6, cox3, nad3, nad4l, nad4, nad5, nad6, cob), two rRNAs (rrnS, rrnL), twenty-two tRNAs (trnF, trnV, trnL2, trnI, trnQ, trnM, trnW, trnA, trnN, trnC, trnY, trnS2, trnD, trnK, trnG, trnR, trnH, trnS1, trnL1, trnE, trnT, trnP), and two non-coding regions of OL and CR are listed in the rows with standard abbreviations, respectively.
The nucleotide compositions of the complete mitochondrial genome for S. kozlovi were 29.59% for A, 25.42% for T, 17.94% for G, and 27.05% for C (Table 3). The nucleotide composition analysis also showed that the A + T content (55.01%) was greater than the G + C content (44.99%) for the mitochondrial genome and the A + T contents in different genes such as PCGs (54.27%), tRNAs (54.80%), rRNAs (54.70%), and CR (66.87%) were higher than G + C content, indicating a bias towards A + T content in the nucleotide composition of S. kozlovi (Table 3). Additionally, the AT-skew had a positive value (AT-skew = 0.08) and the GC-skew had a negative value (GC-skew = −0.20) for the complete mitochondrial genome, which implied that A had a higher abundance than T and C had a higher occurrence than G (Table 3).
Table 3.
The nucleotide composition of the complete mitochondrial genome of S. kozlovi. The nucleotide composition of thirteen PCGs, two rRNAs, twenty-two tRNAs, two non-coding regions of OL and CR, and whole genome are listed in the rows.
3.2. Protein-Coding Genes and Codon Usage
The results of this study showed that the complete mitochondrial genome of S. kozlovi had 13 PCGs with a total length of 11,415 bp accounting for 68.83% of the mitochondrial genome (Table 3). The PCGs consisted of seven NADH dehydrogenases (nad1, nad2, nad3, nad4, nad4l, nad5, nad6), three cytochrome c oxidases (cox1, cox2, cox3), two ATP synthases (atp6 and atp8), and one cytochrome b (cob) (Figure 1). Moreover, 12 PCGs were encoded on the light strand (L), while only nad6 was encoded on the heavy strand (H). The nucleotide compositions of the PCGs were as follows: A = 27.03%, T = 27.24%, G = 17.88%, and C = 27.85%. Both the AT-skew and GC-skew values of the PCGs were found to be negative: AT-skew = −0.003 and GC-skew = −0.22, indicating that there was more T and C in the majority of PCGs (Table 3).
The 13 PCGs of the mitochondrial genome totally encoded 3653 amino acids. All the PCGs used the ATG or GTG as the start codon, where 12 of the 13 PCGs had a start codon of ATG and only the codon for cox1 started with GTG (Table 2). The stop codons were represented by TAA, TAG or single T, where three PCGs (cox2, nad4, cob) were terminated by the stop codon of single T, three PCGs (nad2, nad3, atp8) employed TAG as a stop codon, and the remaining six PCGs (nad1, nad4l, nad5, nad6, cox1, cox3, atp6) were terminated by the TAA stop codon (Table 2). The relative synonymous codon usage (RSCU) values for the 13 PCGs are visualized in Figure 2. The results of RSCU for S. kozlovi suggested that the most commonly used codons were CGA (Arg), CTA (Leu), TCA (Ser), CCA (Pro), ACA (Thr), and we can also conclude that the codon usage of PCGs was biased toward amino acids encoded by A-rich and C-rich codons.
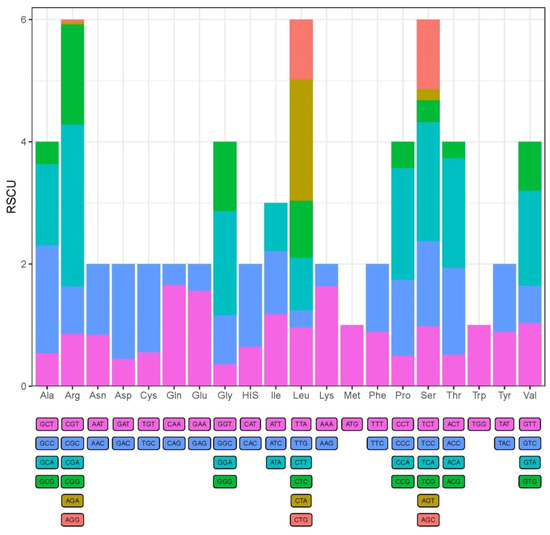
Figure 2.
The relative synonymous codon usage (RSCU) for PCGs in the complete mitochondrial genome of S. kozlovi. The different colors represent different codon families corresponding to amino acids.
3.3. Transfer RNAs, Ribosomal RNAs, and Noncoding Regions
The complete mitochondrial genome of S. kozlovi contained twenty-two transfer RNAs (tRNAs), two ribosomal RNAs (rRNAs), and two non-coding regions (OL and CR). The tRNAs ranged in size from 67 to 76 bp with a total length of 1562 bp (Table 2). It is also clear that eight tRNAs were located on the light strand (L), while the other fourteen tRNAs were encoded on the heavy strand (H). The tRNAs also had an A + T bias so that the A + T content (54.80%) was higher than the G + C content (45.20%) (Table 3). And the tRNAs showed positive values for AT-skew (0.03) and GC-skew (0.05), indicating that the tRNAs were slightly biased towards A and G (Table 3). The inferred secondary structures of the tRNAs supported that 21 of the 22 tRNAs can be folded into the typical cloverleaf secondary structure, which is made up of four domains (AA stem, D arm, AC arm, and T arm), but the other tRNAs of trnaS1 (Ser) was found to lack the D arm in its secondary structure (Figure 3). Additionally, the results also showed that the typical G-C and A-U base pairs were presented in all of the tRNAs. But some mismatched base pairs were also identified in the base pairs, for example 15 tRNAs including trnF, trnL2, trnM, trnW, trnA, trnC, trnY, trnS2, trnD, trnK, trnG, trnH, trnS1, trnE, trnP had G-U or U-G base pairs, and the U-U base pairs appeared in trnF, trnQ, and trnN (Figure 3).
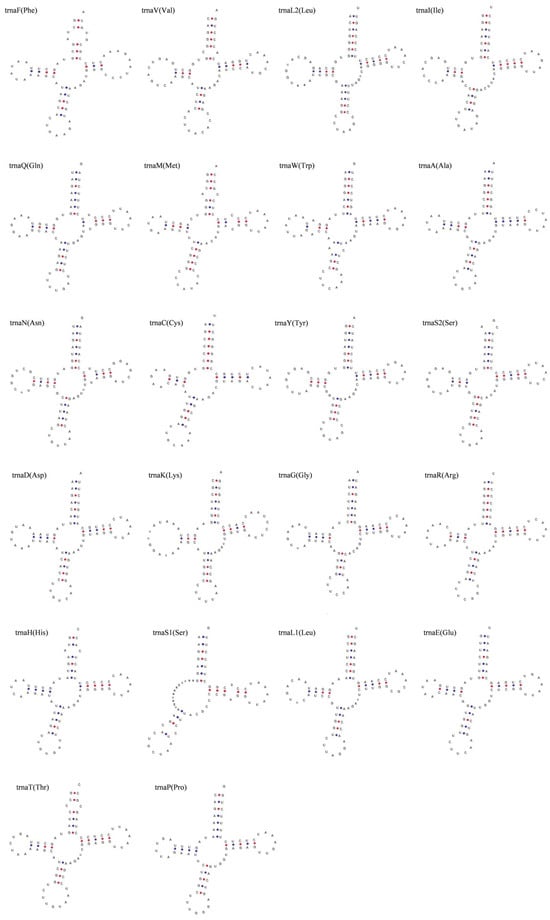
Figure 3.
The inferred secondary structures of tRNAs in the complete mitochondrial genome of S. kozlovi. The 22 tRNA genes were labeled with standard abbreviations in the top left and the corresponding amino acids for translocation were listed in parenthesis, respectively.
The two rRNAs of rrnS and rrnL were both encoded on the heavy strand (H) with lengths of 954 bp and 1629 bp, respectively (Table 2). The rrnS and rrnL were located between trnF and trnL2 and the trnV was interspersed between rRNAs. The rRNAs also had an A + T bias meaning that the A + T content was 54.70%. The AT-skew value for the rRNAs was positive (AT-skew = 0.26), while the GC-skew value for the rRNAs was slightly negative (GC-skew = −0.06) (Table 3). The non-coding regions of the mitochondrial genome of S. kozlovi mainly included the origin of the light strand (OL) and the control region (CR). The OL was located between trnN and trnC with a length of 32 bp. The CR was a relatively long non-coding region with a length of 821 bp, which was located between trnP and trnF and there was an intergenic spacer of 102 bp between the CR and trnF (Figure 1; Table 2). The OL had a significant G + C bias with a G + C content of 65.62%, while the CR had a significant A + T bias with an A + T content of 66.87% (Table 3). The AT-skew values and GC-skew values for OL and CR were negative, indicating that there was more T and C for OL and CR.
3.4. Phylogenetic Relationships
In this study, we determined the phylogenetic relationships of thirty-three species of Schizothorax with two species of Spinibarbus according to the concatenated nucleotide sequences from thirteen PCGs, twenty-two tRNAs, and two rRNAs from mitochondrial genomes based on ML and BI analyses. As a result, the phylogenetic trees generated by the ML and BI methods had the same topological structure and showed well-supported branches. Thus, the current study only used the phylogenetic tree generated through the ML analysis including posterior probabilities and bootstrap values, as shown in Figure 4. The results of the phylogenetic tree showed that the majority of nodes had high support rates with bootstrap proportions ≥ 85 and posterior probabilities ≥ 0.927. While a few nodes in the branches had relatively low support rates, for instance, nodes in the branches had the lowest support rates with a bootstrap proportion of 44 and a posterior probability of 0.49. The results of phylogenetic tree also suggested that the Schizothorax could be classified into four clades: Clade 1 consisted of three species, Clade 2 consisted of eight species, Clade 3 consisted of six species, and Clade 4 consisted of sixteen species (Figure 4). In addition, the topologies of the phylogenetic tree exhibited that the S. kozlovi belonged to Clade 4 and had a relatively close phylogenetic relationship with S. chongi. Thus, the S. kozlovi and S. chongi formed a sister group, which together formed sister groups to other species such as Schizothorax gulinensis, Schizothorax dolichonema, and Schizothorax lissolabiata in Clade 4 (Figure 4).
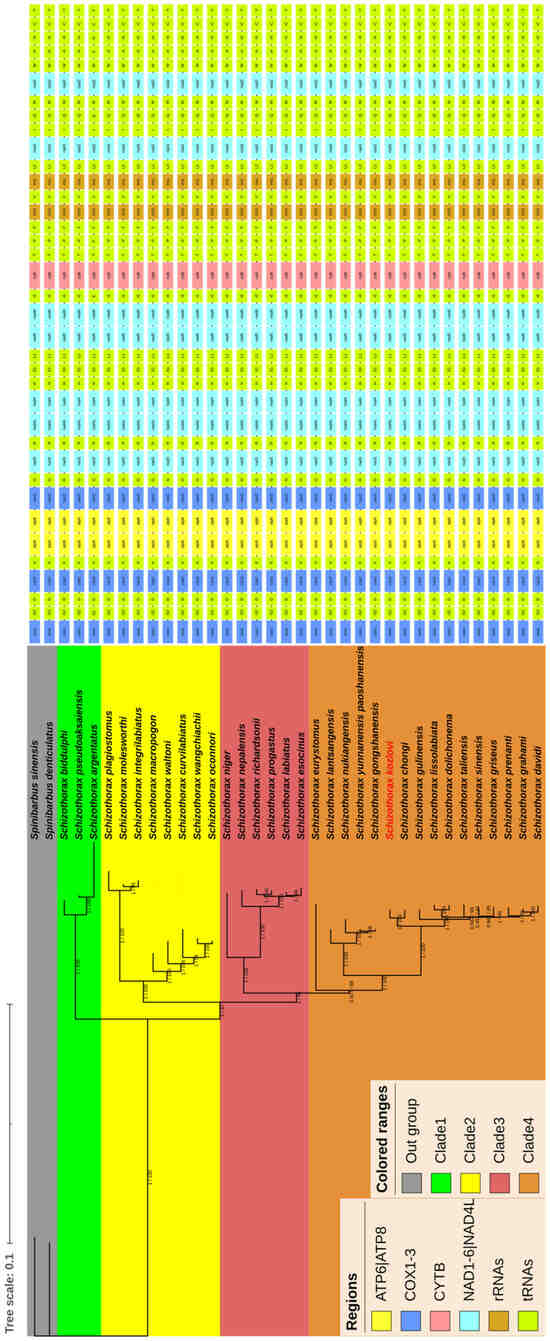
Figure 4.
The phylogenetic tree reconstructed from the nucleotide sequences of thirteen PCGs, twenty-two tRNAs, and two rRNAs using BI and ML methods. Numbers on branches represent posterior probabilities (BI) and bootstrap percentages (ML), respectively. Different colors in regions represent different genes, and gene orders are listed behind the phylogenetic tree. Different colors in the phylogenetic tree represent outgroups and clades. The species of S. kozlovi determined in the present study is marked in red.
4. Discussion
In this study, the characterization of the complete mitochondrial genome of S. kozlovi was described for the first time. Our results indicated that the mitochondrial genome of S. kozlovi is a closed circular structure with a total size of 16,585 bp, which is similar to other reported species of Schizothorax such as Schizothorax davidi, Schizothorax argentatus, and Schizothorax prenanti. Wang et al. [50] suggested that the size of the mitochondrial genome length in closely related species may be caused by variations in the tandem repeat elements within the CR which can explain the gene overlaps. In addition, we also found that the mitochondrial genome of S. kozlovi was composed of thirty-seven genes (thirteen PCGs, twenty-two tRNAs, two rRNAs) and an OL and a CR; the genome organization and composition of which were in accordance with those of most of fish species reported previously [51,52,53]. The AT and GC skews are a measure of asymmetry in the nucleotide composition of the mitochondrial genome. In this study, the A + T content was higher than the G + C content; A had a higher abundance than T so AT-skew was 0.08, and C had a higher occurrence than G so GC-skew was −0.20. Numerous studies have shown that almost all postnatal animals including fishes are characterized by AT bias, indicating that the AT-rich regions might represent the origin of replication and be more easily changed in evolution [54,55]. Generally, the strand skew biases were found to have a negative AT skew and positive GC skew. But, we summarized that the variation in AT and GC skews for different species may change slightly, which can be used as reference evidence for assessing the phylogenetic status and relationships of identified species.
Moreover, it was found that the start codons for 13 PCGs were ATG or GTG, which is consistent with the fish species of Schizothorax wangchiachii and Schizothorax macropogon in Schizothorax [56,57]. In this study, the majority of PCGs were represented by complete stop codons of TAA or TAG. Specifically, three PCGs (cox2, nad4, cob) exhibited incomplete stop codons of single T. Zhang et al. [58] reported that the incomplete stop codons in some PCGs were a common feature of most species of vertebrates. But we also found that there are differences in the incomplete stop codons between species from distinct taxa. For example, Mar-Silva [51] analyzed the mitochondrial genome of Ophisternon infernale and found that seven PCGs had incomplete stop codons for atp6, nad5 used TA as a stop codon, and cox2, cox3, nad4, nad5, and cob employed a single T as the stop codon. However, it is assumed that the incomplete stop codons of TA or T can be modified to TAA via post-transcriptional polyadenylation [59]. The relatively synonymous codon usage (RSCU) can represent the characteristics of codon usage bias in the mitochondrial genome. The results of RSCU analysis for PCGs of S. kozlovi in the current study showed that the most frequently used codons were CGA, CTA, TCA, CCA, and ACA, which encoded the amino acids for Arg, Leu, Ser, Pro, and Thr, respectively. Previous studies have shown that the codon usage relating to functional gene expression and protein sequence encoding might be correlated with natural selection for species [60]. The S. kozlovi is a typical fish species adapted to plateau habitats, thus the codon usage analysis of PCGs in mitochondrial genome can provide an important basis for understanding the origin and evolution of Schizothorax.
The results also showed that, with the exception of trnS1 (Ser) which lacked the D arm, all of the other tRNAs exhibited a canonical cloverleaf secondary structure. Generally, the secondary structure is essential for tRNAs to be associated with stability and function, and changing or removing key parts of the secondary structure of tRNAs might potentially affect amino acid recognition and protein biosynthesis [61]. Yet several studies have reported that trnS1 from most species of vertebrates lacked a D arm and the effects of the loss of D arm on function could be compensated for by other interactions [62,63]. Additionally, our study indicated that the stems of secondary cloverleafs for tRNAs included mostly normal base pairs and some mismatched base pairs. Specifically, trnF, trnL2, trnM, trnW, trnA, trnC, trnY, trnS2, trnD, trnK, trnG, trnH, trnS1, trnE, trnP had G-U or U-G base pairs, and trnF, trnQ, trnN had U-U base pairs. According to Varani and McClain [64], the G-U and U-U wobble base pair is the most common non-Watson–Crick base pair, and the G-U or U-U base pairs have a thermodynamic stability comparable to that of Watson–Crick base pairs and are nearly isomorphic to them, so they would be likely to be the substitutions for a G-C or A-U base pair. The non-coding regions of the mitochondrial genome of S. kozlovi mainly included the origin of the light strand (OL) and the control region (CR), which were closely related to the initiation of the replication and transcription of the mitochondrial genome. Similarly to other fish species, the OL had a relatively short length and usually folded into a hairpin secondary structure [65,66]. The CR in the mitochondrial genome of S. kozlovi had the highest A + T content (66.87%), indicating that the sequence contained AT-rich regions. Previous studies have shown that the CR is mostly a tandem repeat sequence and determines the replication and transcription of the mitochondrial genome. The CR is also considered to be under less selective pressure, and it can, therefore, replicate more efficiently and keep the most rapid evolutionary rate [67,68]. Hence, we can infer that the tandem repeat sequence of different copies as well as the insertion or deletion of gene fragments in the CR might be responsible for variations in the molecular size of the mitochondrial genome of Schizothorax.
In this study, we reconstructed a phylogenetic tree for Schizothorax based on mitochondrial genomes. The results provided some insights into the phylogenetic status of S. kozlovi and its phylogenetic relationships with of Schizothorax. Previously, Rustam et al. [56] conducted a study on the phylogenetic relationships of Schizothoracinae based on 13 PCGs from the mitochondrial genome, and classified Schizothorax as a primitive taxon. Wang et al. [22] inferred the phylogenetic relationships using the 13 PCGs for 28 species and found that 13 species of Schizothorax were clustered into a clade. However, these studies did not provide specific analyses of the phylogenetic relationships of Schizothorax and the species of Schizothorax contained in the phylogenetic analyses were incomplete. In present study, the results supported that the S. kozlovi was closely related to S.chongi, which is consistent with the study by Rustam et al. [56]. Moreover, the topologies of phylogenetic tree divided the Schizothorax into four clades and did not support classification based on morphology which suggested that the Schizothorax can be divided into two subgenera of Schizothorax and Racoma, or into three groups. This result is similar to He and Chen’s [25] analyses of the phylogenetic relationships of Schizothorax based on a single mark of Cyt b. He and Chen [25] and Briolay et al. [69] also reported that adaptive evolution and interbreeding of closely related species can also have impacts on inferring phylogenetic relationships in Schizothorax. The results reminded us that it is easy to be biased in the classification of Schizothorax based solely on morphological or phylogenetic methods, and it is also not sufficiently accurate to infer phylogenetic relationships from a single or a few marks. Therefore, more samples and more sequence data are required to reconstruct a more reliable phylogenetic framework and should be combined with morphological features to accurately determine the status and relationships among species of Schizothorax.
5. Conclusions
The present study reported the complete mitochondrial genome of S. kozlovi for the first time. The mitochondrial genome had a total size of 16,585 bp and contained thirteen PCGs, twenty-two tRNAs, two rRNAs, and two non-coding regions for OL and CR. The Schizothorax could be classified into four clades according to phylogenetic tree, which did not support the classification which divided Schizothorax into two subgenera or three groups based on morphology. In addition, the topologies of the phylogenetic tree showed that the S. kozlovi and S. chongi formed a sister group. The results of the present study have provided basic data and references for the management and conservation of S. kozlovi and Schizothorax.
Author Contributions
Conceptualization, Q.Q. and Y.Z.; methodology, Q.Q. and L.C.; software, Q.Q., J.X. and L.C.; investigation, Q.Q., Y.Z. and F.Z.; writing—original draft preparation, Q.Q.; writing—review and editing, Q.Q., J.X. and Y.Z.; funding acquisition, Q.Q. and Y.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Sichuan, grant number 2022NSFSC1730, 2022NSFSC1646, and the Doctoral Research Launch Special Project of China West Normal University, grant number 21E034.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of China West Normal University (protocol code 2024LLSC0004 and approved on 5 January 2024).
Informed Consent Statement
Not applicable.
Data Availability Statement
Data have been submitted to the GenBank database with accession number OR416862.1 and will be available upon request.
Acknowledgments
We thank Ling Mao, Xin Liu, and other colleagues for the assistance in sample collection and experiments.
Conflicts of Interest
Author L.C. was employed by the company of Powerchina Chengdu Engineering Corporation Limited. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Goodsell, D.S. Mitochondrion. Biochem. Mol. Biol. Educ. 2010, 38, 134–140. [Google Scholar] [CrossRef]
- Garesse, R.; Vallejo, C.G. Animal mitochondrial biogenesis and function: A regulatory cross-talk between two genomes. Gene 2001, 263, 1–16. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Hwang, D.S.; Lee, W.O.; Lee, J.S. Complete mitochondrial genome of the freshwater gudgeon, Pseudopungtungia nigra (Cypriniformes, Gobioninae). Mitochondrial DNA 2014, 25, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Park, J.; Xi, H.; Lee, G.S.; Kim, I.; Park, J. The complete mitochondrial genome of Ricania speculum (Walker, 1851) (Hemiptera: Ricaniidae): Investigation of intraspecific variations on mitochondrial genome. Mitochondrial DNA Part B 2020, 5, 3814–3816. [Google Scholar] [CrossRef]
- Galtier, N.; Nabholz, B.; Glémin, S.; Hurst, G.D. Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Mol. Ecol. 2009, 18, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.P.; Miya, M.; Mabuchi, K.; Nishida, M. Structure and variation of the mitochondrial genome of fishes. BMC Genom. 2016, 17, 719. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Hasegawa, M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol. Biol. Evol. 1998, 15, 1600–1611. [Google Scholar] [CrossRef]
- Zardoya, R.; Meyer, A. Cloning and characterization of a microsatellite in the mitochondrial control region of the African side-necked turtle, Pelomedusa subrufa. Gene 1998, 216, 149–153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caccone, A.; Gentile, G.; Burns, C.E.; Sezzi, E.; Bergman, W.; Ruelle, M.; Saltonstall, K.; Powell, J.R. Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectotherm, Galápagos tortoises. Mol. Phylogenet. Evol. 2004, 31, 794–798. [Google Scholar] [CrossRef]
- Keith, P.; Lord, C.; Lorion, J.; Watanabe, S.; Tsukamoto, K.; Couloux, A.; Dettai, A. Phylogeny and biogeography of Sicydiinae (Teleostei: Gobiidae) inferred from mitochondrial and nuclear genes. Mar. Biol. 2011, 158, 311–326. [Google Scholar] [CrossRef]
- Shao, R.; Barker, S.C. Mitochondrial genomes of parasitic arthropods: Implications for studies of population genetics and evolution. Parasitology 2007, 134, 153–167. [Google Scholar] [CrossRef]
- Song, N.; Lin, A.; Zhao, X. Insight into higher-level phylogeny of Neuropterida: Evidence from secondary structures of mitochondrial rRNA genes and mitogenomic data. PLoS ONE 2018, 13, e0191826. [Google Scholar] [CrossRef]
- Ding, R.H. The Fishes of Sichuan, China; Sichuan Publishing House of Science and Technology: Chengdu, China, 1994. [Google Scholar]
- Lin, P.C.; Miao, Z.G.; Gao, X.; Liu, H.Z. Length-weight relationships of 11 fish species from the upper Jinsha River, China. J. Appl. Ichthyol. 2015, 31, 223–224. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, J.; Wang, Y.; Zhang, E.; Zhang, Y. Red list of China’s vertebrates. Biodiv. Sci. 2016, 24, 500–551. [Google Scholar]
- Chen, Y.; Luo, Q. The fecundity of Schizothorax kozlovi from Wu River. Zool. Res. 1995, 16, 324–342. [Google Scholar]
- Zhang, X.; Dai, Y. Feeding habits and resources protection of Schizothorax kozlovi. J. Hydroecol. 2010, 32, 110–114. [Google Scholar] [CrossRef]
- He, Y.; Wu, X.; Zhu, Y.; Li, H.; Li, X.; Yang, D. Effect of rearing temperature on growth and thermal tolerance of Schizothorax (Racoma) kozlovi larvae and juveniles. J. Therm. Biol. 2014, 46, 24–30. [Google Scholar] [CrossRef]
- He, J.; He, Z.; Yang, D.; Ma, Z.; Chen, H.; Zhang, Q.; Deng, F.; Ye, L.; Pu, Y.; Zhang, M.; et al. Genetic variation in Schizothorax kozlovi Nikolsky in the upper reaches of the Chinese Yangtze River based on genotyping for simplified genome sequencing. Animals 2022, 12, 2181. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Gong, J.; Wu, X.; Zhu, Y.; Yang, D. Population structure of wild Schizothorax kozlovi in the upper Yangtze River based on mtDNA and stable isotopes, and their relationship with ambient temperature. Fishes 2022, 7, 292. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, P.; Dai, Y.; Xu, D.; Dong, Y.; Huang, Z. The complete mitochondrial genome of a new species of the genus Schizothorax from Sichuan, China (Cypriniformes: Cyprinidae). Mitochondrial DNA Part B 2023, 8, 1356–1359. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.F.; Wu, C.Z. The Fishes of the Qinghai-Xizang Plateau; Sichuan Publishing House of Science and Technology: Chengdu, China, 1991. [Google Scholar]
- Wu, Y.F. Systematic studies on the cyprinid fishes of the subfamily schizothoracinae from China. Acta Biologica Plateau Sinica 1984, 3, 119–140. [Google Scholar]
- He, D.; Chen, Y. Biogeography and molecular phylogeny of the genus Schizothorax (Teleostei: Cyprinidae) in China inferred from cytochrome b sequences. J. Biogeogr. 2006, 33, 1448–1460. [Google Scholar] [CrossRef]
- He, D.K.; Chen, Y.F.; Chen, Y.Y.; Chen, Z.M. Molecular phylogeny of the specialized schizothoracine fishes (Teleostei: Cyprinidae), with their implications for the uplift of the Qinghai-Tibetan Plateau. Chin. Sci. Bull. 2004, 49, 39–48. [Google Scholar] [CrossRef]
- Cao, W.X.; Chen, Y.Y.; Wu, Y.F. Origin and evolution of Schizothorax and their relationship with the uplift of the Qinghai-Xizang Plateau. In Ages, Amplitudes and Form Problems during the Uplift of the Qinghai-Xizang Plateau; The Qinghai-Tibet Plateau Comprehensive Scientific Expedition from Chinese Academy of Sciences, Ed.; Science Press: Beijing, China, 1981. [Google Scholar]
- Wu, Y.F.; Tan, Q.J. Characteristics of the fish-fauna of the characteristics of Qinghai-Xizang Plateau and its geological distribution and formation. Acta Zool. Sin. 1991, 37, 135–152. [Google Scholar]
- Chen, Y.F.; Cao, W.X. Schizothoracinae. In Fauna Sinica, Osteichthyes, Cypriniformes II; Yue, P.Q., Ed.; Science Press: Beijing, China, 2000. [Google Scholar]
- Heckel, J.J. Fische aus Caschmir; P.P. Mechitaristen: Wien, Austria, 1838. [Google Scholar]
- Chen, Y.Y. Fauna Sinica, Osteichthyes, Cypriniformes II; Science Press: Beijing, China, 1998. [Google Scholar]
- Bartlett, S.E.; Davidson, W.S. Identification of Thunnus tuna species by the polymerase chain reaction and direct sequence analysis of their mitochondrial cytochrome b genes. Can. J. Fish. Aquat. Sci. 1991, 48, 309–317. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data; Babraham Institute: Cambridge, UK, 2013. [Google Scholar]
- Duan, X.; Dong, X.; Li, J.; Lü, J.; Guo, B.; Xu, K.; Ye, Y. The complete mitochondrial genome of Pilumnopeus Makianus (Brachyura: Pilumnidae), novel gene rearrangements, and phylogenetic relationships of Brachyura. Genes 2022, 13, 1943. [Google Scholar] [CrossRef]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2016, 45, e18. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Yang, T.; Xu, G.; Gu, B.; Shi, Y.; Mzuka, H.L.; Shen, H. The complete mitochondrial genome sequences of the Philomycus bilineatus (Stylommatophora: Philomycidae) and phylogenetic analysis. Genes 2019, 10, 198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Xiang, C.-Y.; Gao, F.; Jakovlić, I.; Lei, H.-P.; Hu, Y.; Zhang, H.; Zou, H.; Wang, G.-T.; Zhang, D. Using PhyloSuite for molecular phylogeny and tree-based analyses. iMeta 2023, 2, e87. [Google Scholar] [CrossRef]
- Wang, I.C.; Lin, H.-D.; Liang, C.-M.; Huang, C.-C.; Wang, R.-D.; Yang, J.-Q.; Wang, W.-K. Complete mitochondrial genome of the freshwater fish Onychostoma lepturum (Teleostei, Cyprinidae): Genome characterization and phylogenetic analysis. ZooKeys 2020, 1005, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Mar-Silva, A.F.; Arroyave, J.; Díaz-Jaimes, P. The complete mitochondrial genome of the Mexican-endemic cavefish Ophisternon infernale (Synbranchiformes, Synbranchidae): Insights on patterns of selection and implications for synbranchiform phylogenetics. ZooKeys 2022, 1089, 1–23. [Google Scholar] [CrossRef]
- Mao, L.; Zeng, Y.; Li, J.; Zhang, F.; Liu, Y.; Gong, J. The complete mitochondrial genome sequence and phylogenetic analysis of Gnathopogon herzensteini (Cypriniformes, Cyprinidae, Gobioninae). Biologia 2021, 76, 1087–1094. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Xu, Q. Complete mitochondrial genome of Schizothorax gongshanensis (Cypriniformes: Cyprinidae). Mitochondrial DNA Part B 2016, 1, 528–529. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, S.; Kumazawa, Y.; Araki, T.; Himeno, H.; Miura, K.-I.; Watanabe, K. Strand-specific nucleotide composition bias in echinoderm and vertebrate mitochondrial genomes. J. Mol. Evol. 1991, 32, 511–520. [Google Scholar] [CrossRef]
- Cui, L.; Cao, R.; Dong, Y.; Gao, X.; Cen, J.; Lu, S. The first complete mitochondrial genome of the flathead Cociella crocodilus (Scorpaeniformes: Platycephalidae) and the phylogenetic relationships within Scorpaeniformes based on whole mitogenomes. Genes 2019, 10, 533. [Google Scholar] [CrossRef]
- Rustam, D.; Yuan, X.; Zhang, Q.; Han, J. Study on the phylogeny of Schizothoracids based on complete mitochondrial genome. J. Fish. Sci. China 2022, 29, 781–791. [Google Scholar] [CrossRef]
- Chen, Y.; Cheng, Q.; Qiao, H.; Zhu, Y.; Chen, W.; Ren, G. The complete mitochondrial genome sequence of Schizothorax wangchiachii (Cypriniformes: Cyprinidae). Mitochondrial DNA 2013, 24, 353–355. [Google Scholar] [CrossRef]
- Zhang, H.; Fang, W.; Zhao, X.; Jiang, X.; Stroiński, A.; Qin, D. Comparative analysis of the complete mitochondrial genomes of five species of Ricaniidae (Hemiptera: Fulgoromorpha) and phylogenetic implications. Biology 2022, 11, 92. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Ben Slimen, H.; Awadi, A.; Tolesa, Z.G.; Knauer, F.; Alves, P.C.; Makni, M.; Suchentrunk, F. Positive selection on the mitochondrial ATP synthase 6 and the NADH dehydrogenase 2 genes across 22 hare species (genus Lepus). J. Zool. Syst. Evol. Res. 2018, 56, 428–443. [Google Scholar] [CrossRef]
- Pons, J.; Bauzà-Ribot, M.M.; Jaume, D.; Juan, C. Next-generation sequencing, phylogenetic signal and comparative mitogenomic analyses in Metacrangonyctidae (Amphipoda: Crustacea). BMC Genom. 2014, 15, 566. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Brown, W.M.; Boore, J.L. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc. Natl. Acad. Sci. USA 2000, 97, 13738–13742. [Google Scholar] [CrossRef]
- Li, T.; Yang, J.; Li, Y.; Cui, Y.; Xie, Q.; Bu, W.; Hillis, D.M. A mitochondrial genome of Rhyparochromidae (Hemiptera: Heteroptera) and a comparative analysis of related mitochondrial genomes. Sci. Rep. 2016, 6, 35175. [Google Scholar] [CrossRef] [PubMed]
- Varani, G.; McClain, W.H. The G·U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef]
- Mao, M.; Gu, J.; Liu, R.; Chen, Y.; Wu, L.; Jiang, Z.; Jiang, J. Analysis of complete mitochondrial genome sequences of Gadus macrocephalus. Acta Hydrobiol. Sin. 2019, 43, 17–26. [Google Scholar] [CrossRef]
- Yu, X.; Tan, W.; Zhang, H.; Jiang, W.; Gao, H.; Wang, W.; Liu, Y.; Wang, Y.; Tian, X. Characterization of the complete mitochondrial genome of Harpalus sinicus and its implications for phylogenetic analyses. Genes 2019, 10, 724. [Google Scholar] [CrossRef]
- Shao, R.; Barker, S.C.; Mitani, H.; Aoki, Y.; Fukunaga, M. Evolution of duplicate control regions in the mitochondrial genomes of Metazoa: A case study with Australasian Ixodes Ticks. Mol. Biol. Evol. 2004, 22, 620–629. [Google Scholar] [CrossRef]
- Zheng, C.; Nie, L.; Wang, J.; Zhou, H.; Hou, H.; Wang, H.; Liu, J. Recombination and evolution of duplicate control regions in the mitochondrial genome of the Asian big-headed turtle, Platysternon megacephalum. PLoS ONE 2013, 8, e82854. [Google Scholar] [CrossRef] [PubMed]
- Briolay, J.; Galtier, N.; Brito, R.M.; Bouvet, Y. Molecular phylogeny of Cyprinidae inferred from cytochrome b DNA sequences. Mol. Phylogenet. Evol. 1998, 9, 100–108. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).