

Characterization of Eighty-Eight Single-Nucleotide Polymorphism Markers in the Manila Clam Ruditapes philippinarum Based on High-Resolution Melting (HRM) Analysis


 ,
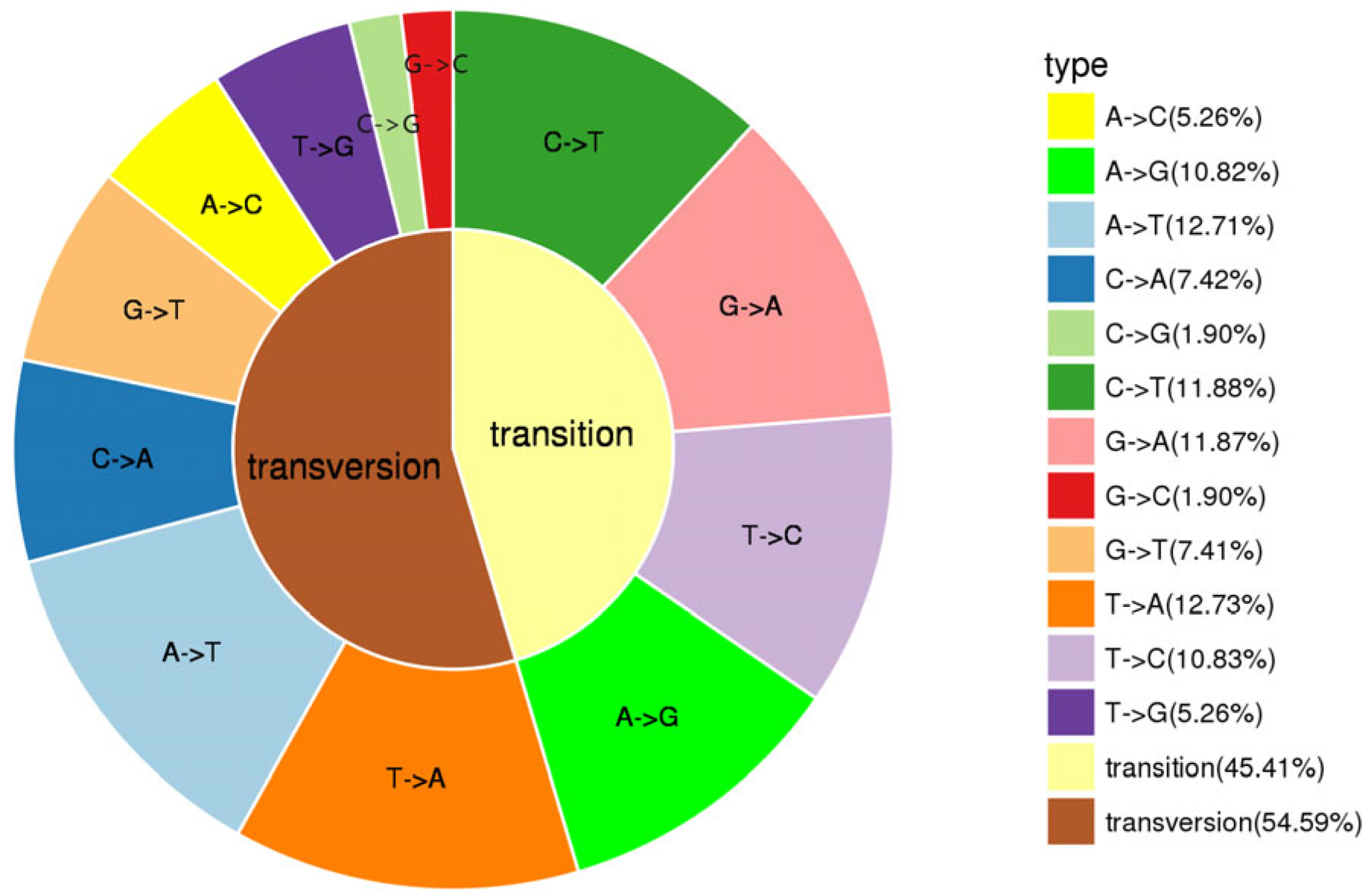
, 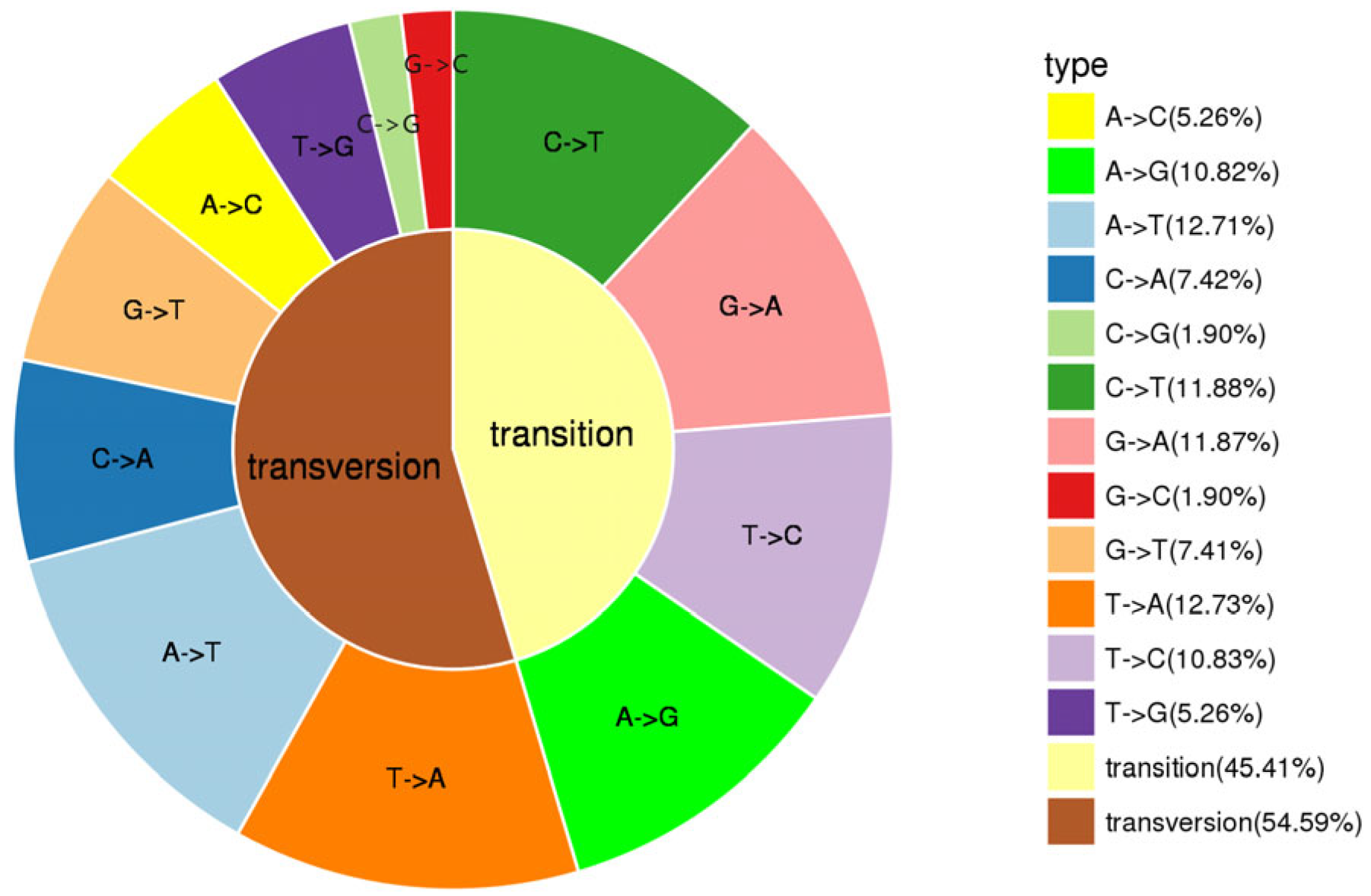{kind=link}
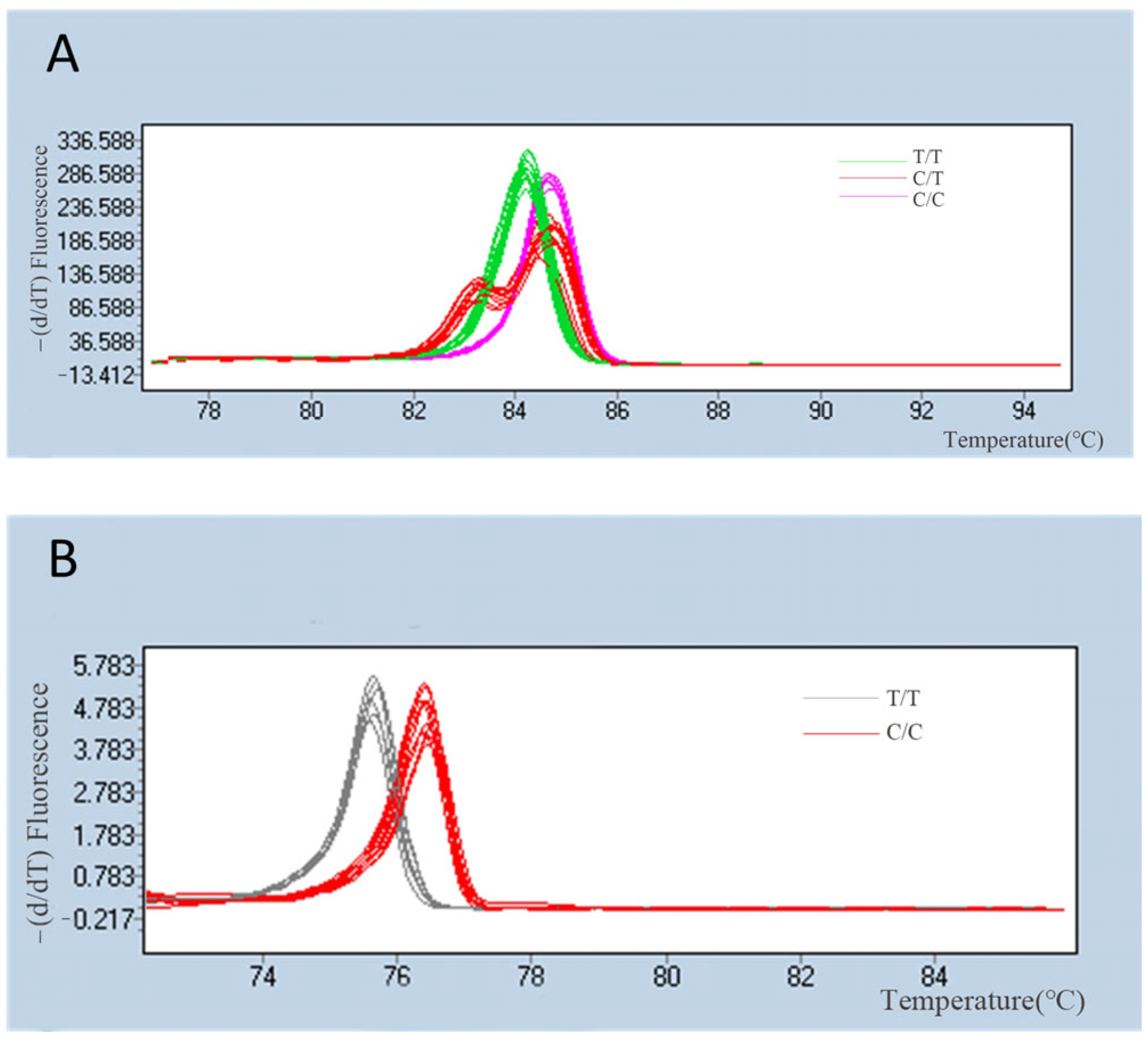
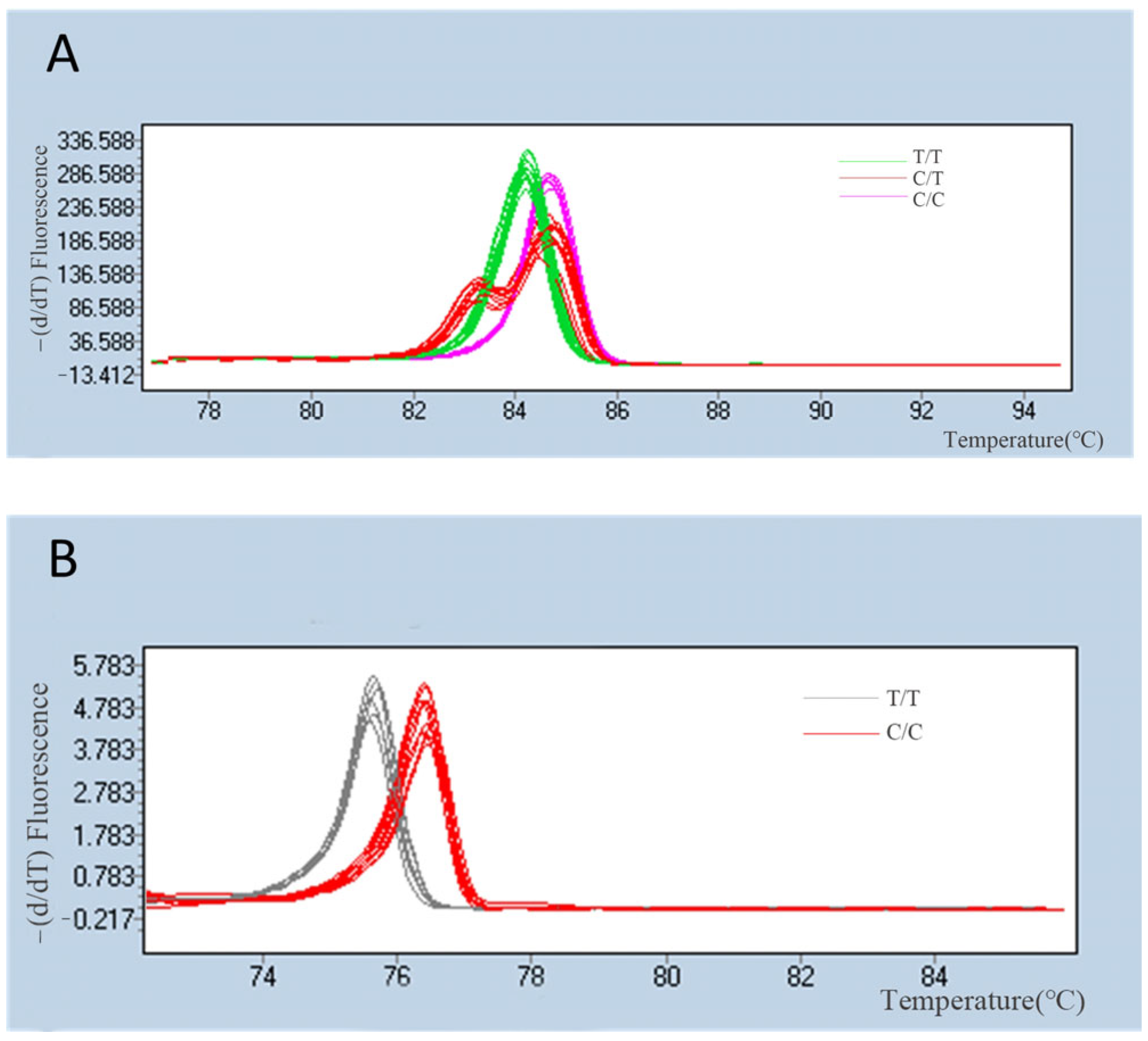{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Sampling
2.2. DNA Extraction and RAD-seq Library Preparation
2.3. Bioinformatics Analysis
2.3.1. Clean Reads Filtering
2.3.2. SNP Identification and Annotation
2.4. Standard Population Genetic Statistics
3. Results
3.1. RAD Data Analysis
3.2. Evaluation of SNP Markers for Population Genomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- FAO. The state of world fisheries and aquaculture 2018–meeting the sustainable development goals. In The State of World Fisheries and Aquaculture; Food and Agriculture Organization of the United Nations: Rome, Italy, 2018. [Google Scholar]
- You, X.; Shan, X.; Shi, Q. Research advances in the genomics and applications for molecular breeding of aquaculture animals. Aquaculture 2020, 526, 735357. [Google Scholar] [CrossRef]
- Robledo, D.; Palaiokostas, C.; Bargelloni, L.; Martínez, P.; Houston, R. Applications of genotyping by sequencing in aquaculture breeding and genetics. Rev. Aquac. 2018, 10, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.C.; Grabherr, M.G.; Chan, Y.F.; Russell, P.; Mauceli, E.; Johnson, J.; Swofford, R.; Pirun, M.; Zody, M.C.; White, S. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 2012, 484, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Chavanne, H.; Janssen, K.; Hofherr, J.; Contini, F.; Haffray, P.; Consortium, A.; Komen, H.; Nielsen, E.E.; Bargelloni, L. A comprehensive survey on selective breeding programs and seed market in the European aquaculture fish industry. Aquac. Int. 2016, 24, 1287–1307. [Google Scholar] [CrossRef]
- Jiao, W.; Fu, X.; Li, J.; Li, L.; Feng, L.; Lv, J.; Zhang, L.; Wang, X.; Li, Y.; Hou, R.; et al. Large-scale development of gene-associated single-nucleotide polymorphism markers for molluscan population genomic, comparative genomic, and genome-wide association studies. DNA Res. 2014, 21, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Guppy, J.L.; Jones, D.B.; Kjeldsen, S.R.; Le Port, A.; Khatkar, M.S.; Wade, N.M.; Sellars, M.J.; Steinig, E.J.; Raadsma, H.W.; Jerry, D.R.; et al. Development and validation of a RAD-Seq target-capture based genotyping assay for routine application in advanced black tiger shrimp (Penaeus monodon) breeding programs. BMC Genom. 2020, 21, 541. [Google Scholar] [CrossRef]
- Bruneaux, M.; Johnston, S.E.; Herczeg, G.; Merilä, J.; Primmer, C.R.; Vasemägi, A. Molecular evolutionary and population genomic analysis of the nine-spined stickleback using a modified restriction-site-associated DNA tag approach. Mol. Ecol. 2013, 22, 565–582. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Puritz, J.B.; Wang, Z.; Proestou, D.; Allen Jr, S.; Small, J.; Verbyla, K.; Zhao, H.; Haggard, J.; Chriss, N. Development and evaluation of high-density SNP arrays for the Eastern oyster Crassostrea virginica. Mar. Biotechnol. 2023, 25, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Palti, Y.; Gao, G.; Liu, S.; Kent, M.; Lien, S.; Miller, M.; Rexroad III, C.; Moen, T. The development and characterization of a 57 K single nucleotide polymorphism array for rainbow trout. Mol. Ecol. Resour. 2015, 15, 662–672. [Google Scholar] [CrossRef]
- Qi, H.; Song, K.; Li, C.; Wang, W.; Li, B.; Li, L.; Zhang, G. Construction and evaluation of a high-density SNP array for the Pacific oyster (Crassostrea gigas). PLoS ONE 2017, 12, e0174007. [Google Scholar] [CrossRef]
- Sun, X.; Shin, G.; Hedgecock, D. Inheritance of high-resolution melting profiles in assays targeting single nucleotide polymorphisms in protein-coding sequences of the Pacific oyster Crassostrea gigas: Implications for parentage assignment of experimental and commercial broodstocks. Aquaculture 2015, 437, 127–139. [Google Scholar] [CrossRef]
- Taylor, C.F. Mutation scanning using high-resolution melting. Biochem. Soc. Trans. 2009, 37, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Kang, Y.; Torres-Jerez, I.; Cheung, F.; Town, C.D.; Zhao, P.X.; Udvardi, M.K.; Monteros, M.J. Genome-wide SNP discovery in tetraploid alfalfa using 454 sequencing and high resolution melting analysis. BMC Genom. 2011, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Radvanszky, J.; Surovy, M.; Nagyova, E.; Minarik, G.; Kadasi, L. Comparison of different DNA binding fluorescent dyes for applications of high-resolution melting analysis. Clin. Biochem. 2015, 48, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Fang, L.; Qiu, M.; Huo, Z.; Yan, X. Population genetics of the Manila clam (Ruditapes philippinarum) in East Asia. Sci. Rep. 2020, 10, 21890. [Google Scholar] [CrossRef]
- Zheng, S.; Zhang, T.; Tu, K.; Li, L.; Liu, Z.; Wu, B.; Zhou, L.; Sun, X. Population Genetics of Manila Clam (Ruditapes philippinarum) in China Inferred from Microsatellite Markers. Biology 2023, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- DOF. China Fisheries Statistic Yearbook; China Agriculture Press: Beijing, China, 2021. [Google Scholar]
- Nie, H.; Yan, X.; Huo, Z.; Jiang, L.; Chen, P.; Liu, H.; Ding, J.; Yang, F. Construction of a high-density genetic map and quantitative trait locus mapping in the manila clam Ruditapes philippinarum. Sci. Rep. 2017, 7, 229. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chiki, L.; Raufaste, N. 4.05, Logiciel sous Windows TM pour la Génétique des Populations; Université de Montpellier II, Laboratoire Génome, Populations, Interactions: Montpellier, France, 2004. [Google Scholar]
- Nie, H.; Liu, L.; Ding, J.; Yan, X. Characterization of fourteen single nucleotide polymorphism markers in the Manila clam (Ruditapes philippinarum). Conserv. Genet. Resour. 2015, 7, 1–4. [Google Scholar] [CrossRef]
- Jin, Y.-L.; Kong, L.-F.; Yu, H.; Li, Q. Development, inheritance and evaluation of 55 novel single nucleotide polymorphism markers for parentage assignment in the Pacific oyster (Crassostrea gigas). Genes Genom. 2014, 36, 129–141. [Google Scholar] [CrossRef]
- Dong, Y.; Jing, Y.; Ruan, W.; Yao, H.; He, L.; Lin, Z. Development and evaluation of a set of 135 EST-SNP markers in the transcriptome dataset of hard clam, Meretrix meretrix. Conserv. Genet. Resour. 2018, 10, 651–658. [Google Scholar] [CrossRef]
- Wang, S.; Sha, Z.; Sonstegard, T.S.; Liu, H.; Xu, P.; Somridhivej, B.; Peatman, E.; Kucuktas, H.; Liu, Z. Quality assessment parameters for EST-derived SNPs from catfish. BMC Genom. 2008, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, Q. Characterization of novel EST-SNP markers and their association analysis with growth-related traits in the Pacific oyster Crassostrea gigas. Aquac. Int. 2017, 25, 1707–1719. [Google Scholar] [CrossRef]
- Conover, D.O.; Clarke, L.M.; Munch, S.B.; Wagner, G.N. Conover DO, Clarke LM, Munch SB, Wagner GN. Spatial and temporal scales of adaptive divergence in marine fishes and the implication for conservation. J. Fish Biol. 2006, 69, 21–47. [Google Scholar] [CrossRef]
- Sun, X.; Li, L.; Wang, X. Development and characterization of 37 SNP markers in the ark shell Scapharca subcrenata using RAD sequencing and high resolution melting analysis. Conserv. Genet. Resour. 2017, 9, 365–368. [Google Scholar] [CrossRef]
- Nie, H.; Niu, H.; Zhao, L.; Yang, F.; Yan, X.; Zhang, G. Genetic diversity and structure of Manila clam (Ruditapes philippinarum) populations from Liaodong peninsula revealed by SSR markers. Biochem. Syst. Ecol. 2015, 59, 116–125. [Google Scholar] [CrossRef]
- Jiqin, L.I.; Zhenmin, B.; Ling, L.I.; Xiaojian, W.; Shi, W.; Xiaoli, H.U. Development of 101 Novel EST-Derived Single Nucleotide Polymor- phism Markers for Zhikong Scallop (Chlamysfarreri). J. Ocean Univ. China 2013, 012, 403–412. [Google Scholar]
- Sauvage, C.; Bierne, N.; Lapègue, S.; Boudry, P. Single Nucleotide polymorphisms and their relationship to codon usage bias in the Pacific oyster Crassostrea gigas. Gene 2007, 406, 13–22. [Google Scholar] [CrossRef]
- Hong-Jun, L.I.; Yu, L.; Kun, X.; Hao, S.U.; Xiang-Gang, G.; Li-Jun, S.; Chong-Bo, H.E. Development of single nucleotide polymorphism markers for blue mussel (Mytilus galloprovincialis) using expressed sequence tags. J. Fish. China 2011, 35, 348–355. [Google Scholar]
- Katju, V.; Bergthorsson, U. Old trade, new tricks: Insights into the spontaneous mutation process from the partnering of classical mutation accumulation experiments with high-throughput genomic approaches. Genome Biol. Evol. 2019, 11, 136–165. [Google Scholar] [CrossRef] [PubMed]
- Song, K. Genomic landscape of mutational biases in the Pacific oyster Crassostrea gigas. Genome Biol. Evol. 2020, 12, 1943–1952. [Google Scholar] [CrossRef] [PubMed]
- Talavera, A.; Soorni, A.; Bombarely, A.; Matas, A.J.; Hormaza, J.I. Genome-Wide SNP discovery and genomic characterization in avocado (Persea americana Mill.). Sci. Rep. 2019, 9, 20137. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, N.; Nagai, S.; Yamaguchi, S.; Lian, C.; Hamaguchi, M. Development of microsatellite markers for the Manila clam Ruditapes philippinarum. Mol. Ecol. Notes 2007, 7, 43–45. [Google Scholar] [CrossRef]
- Wei, D.; Zheng, S.; Wang, S.; Yan, J.; Liu, Z.; Zhou, L.; Wu, B.; Sun, X. Genetic and Haplotype Diversity of Manila Clam Ruditapes philippinarum in Different Regions of China Based on Three Molecular Markers. Animals 2023, 13, 2886. [Google Scholar] [CrossRef]
- Nie, H.-T.; Zhu, D.-P.; Yang, F.; Zhao, L.-Q.; Yan, X.-W. Development and characterization of EST-derived microsatellite makers for Manila clam (Ruditapes philippinarum). Conserv. Genet. Resour. 2014, 6, 25–27. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Chen, Y.; Wu, B.; Zhou, L.; Liu, Z.; Zhang, T.; Sun, X. Characterization of Eighty-Eight Single-Nucleotide Polymorphism Markers in the Manila Clam Ruditapes philippinarum Based on High-Resolution Melting (HRM) Analysis. Animals 2024, 14, 542. https://doi.org/10.3390/ani14040542
Zheng S, Chen Y, Wu B, Zhou L, Liu Z, Zhang T, Sun X. Characterization of Eighty-Eight Single-Nucleotide Polymorphism Markers in the Manila Clam Ruditapes philippinarum Based on High-Resolution Melting (HRM) Analysis. Animals. 2024; 14(4):542. https://doi.org/10.3390/ani14040542
Chicago/Turabian StyleZheng, Sichen, Yancui Chen, Biao Wu, Liqing Zhou, Zhihong Liu, Tianshi Zhang, and Xiujun Sun. 2024. "Characterization of Eighty-Eight Single-Nucleotide Polymorphism Markers in the Manila Clam Ruditapes philippinarum Based on High-Resolution Melting (HRM) Analysis" Animals 14, no. 4: 542. https://doi.org/10.3390/ani14040542
APA StyleZheng, S., Chen, Y., Wu, B., Zhou, L., Liu, Z., Zhang, T., & Sun, X. (2024). Characterization of Eighty-Eight Single-Nucleotide Polymorphism Markers in the Manila Clam Ruditapes philippinarum Based on High-Resolution Melting (HRM) Analysis. Animals, 14(4), 542. https://doi.org/10.3390/ani14040542