

Transcriptome and Proteome Analyses Revealed Differences in JEV-Infected PK-15 Cells in Response to Ferroptosis Agonists and Antagonists

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viral Infection
2.2. Real-Time Quantitative PCR Analysis
2.3. Western Blot Analysis
2.4. Oxidative Stress Analysis
2.5. Transcriptome Sequencing and Differentially Expressed Gene Analysis
2.6. Proteome Sequencing and Analysis of Differentially Expressed Proteins
2.7. Statistical Analysis
3. Results
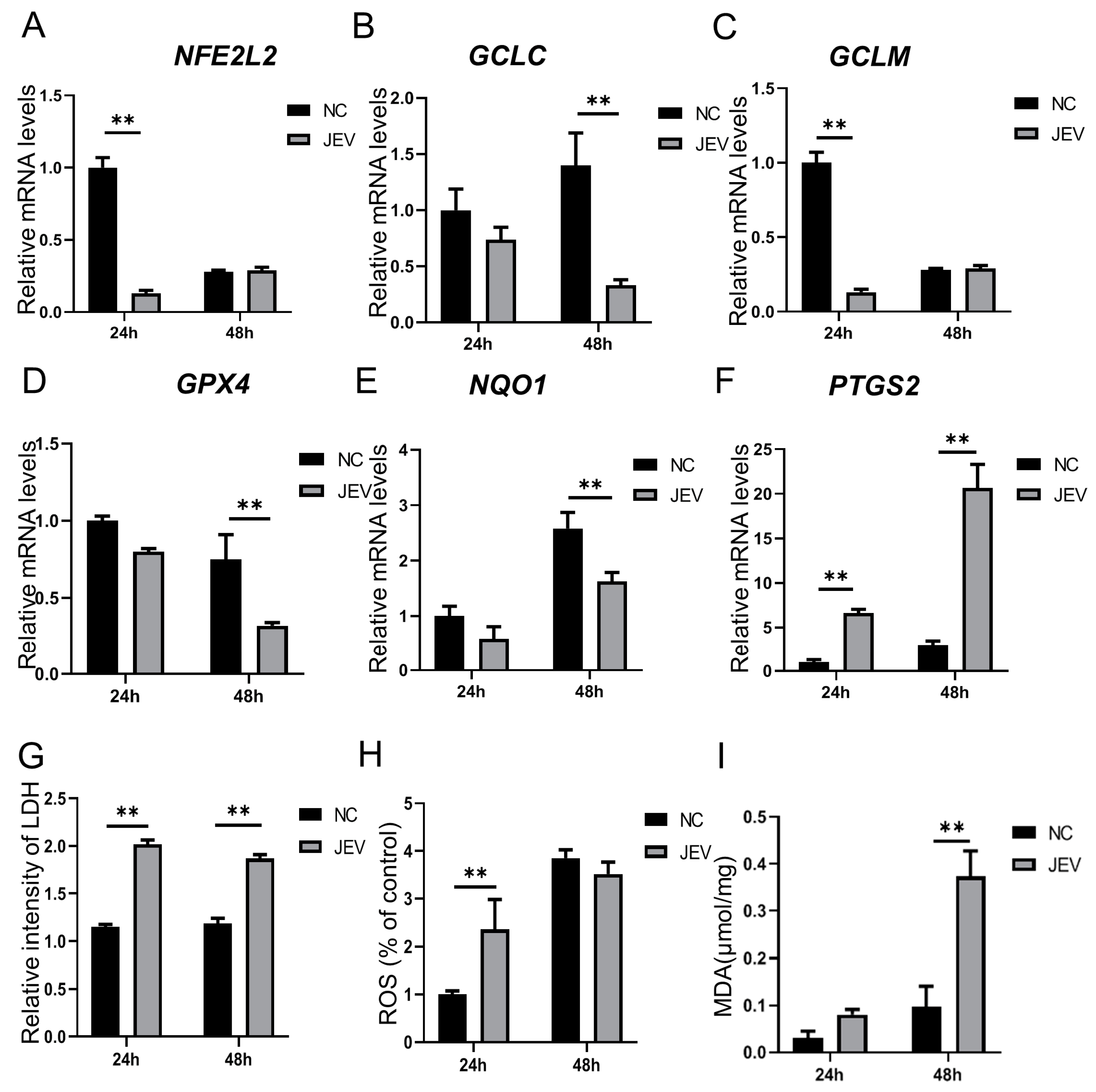
3.1. Ferroptosis-Related Genes and Indicators Were Regulated by JEV Infection in PK-15 Cells
3.2. Effects of Ferroptosis Agonists and Antagonists on JEV Proliferation
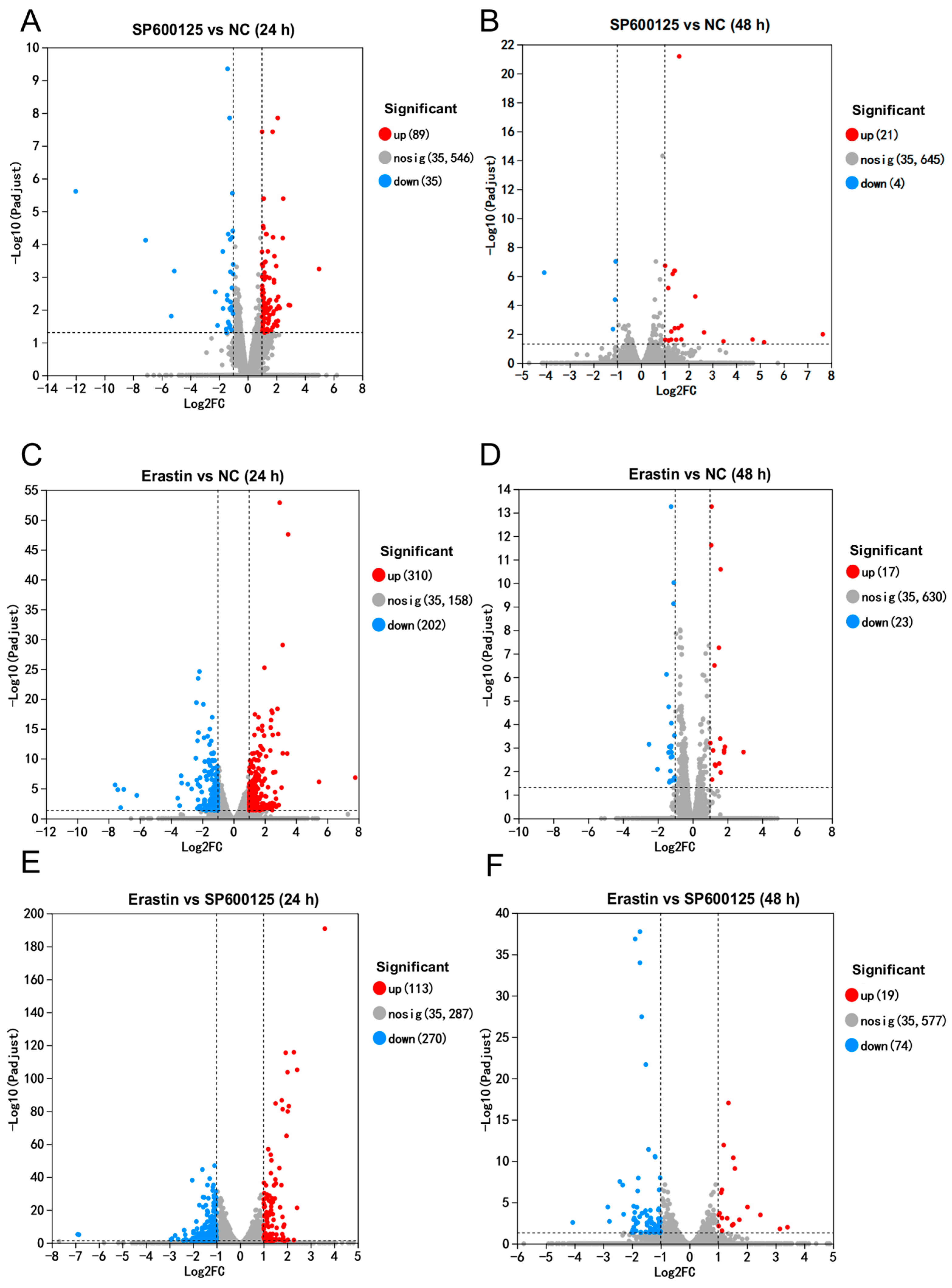
3.3. Differentially Expressed Genes Induced by Ferroptosis Agonist and Antagonist in JEV Infected PK-15 Cells
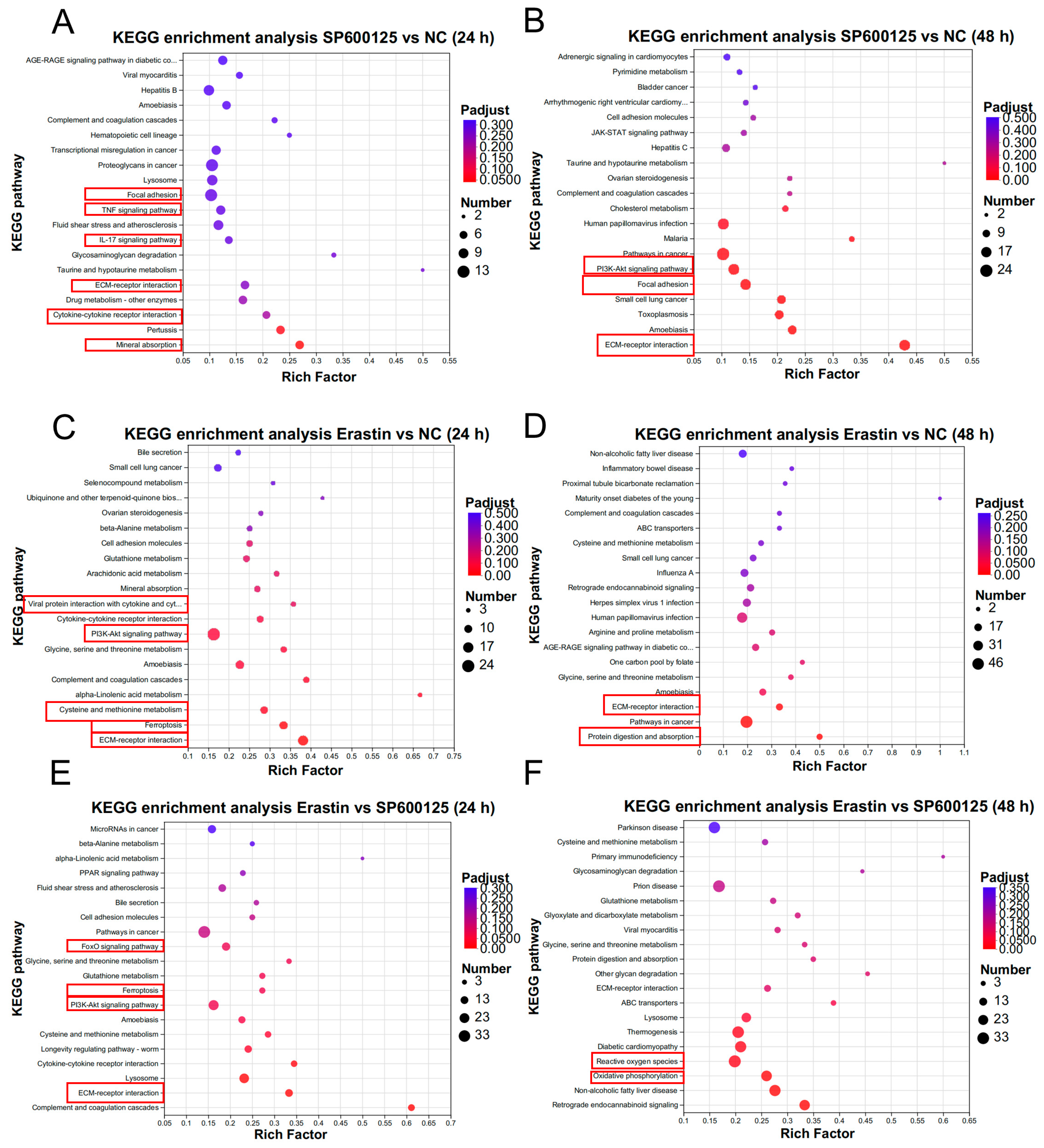
3.4. Gene Expression Pattern Analysis for Ferroptosis Agonist and Antagonist Treatment in JEV-Infected PK-15 Cells
3.5. Differentially Expressed Proteins Induced by Ferroptosis Agonist and Antagonist in JEV-Infected PK-15 Cells
3.6. Protein Expression Pattern Analysis for Ferroptosis Agonist and Antagonist Treatments in JEV-Infected PK-15 Cells
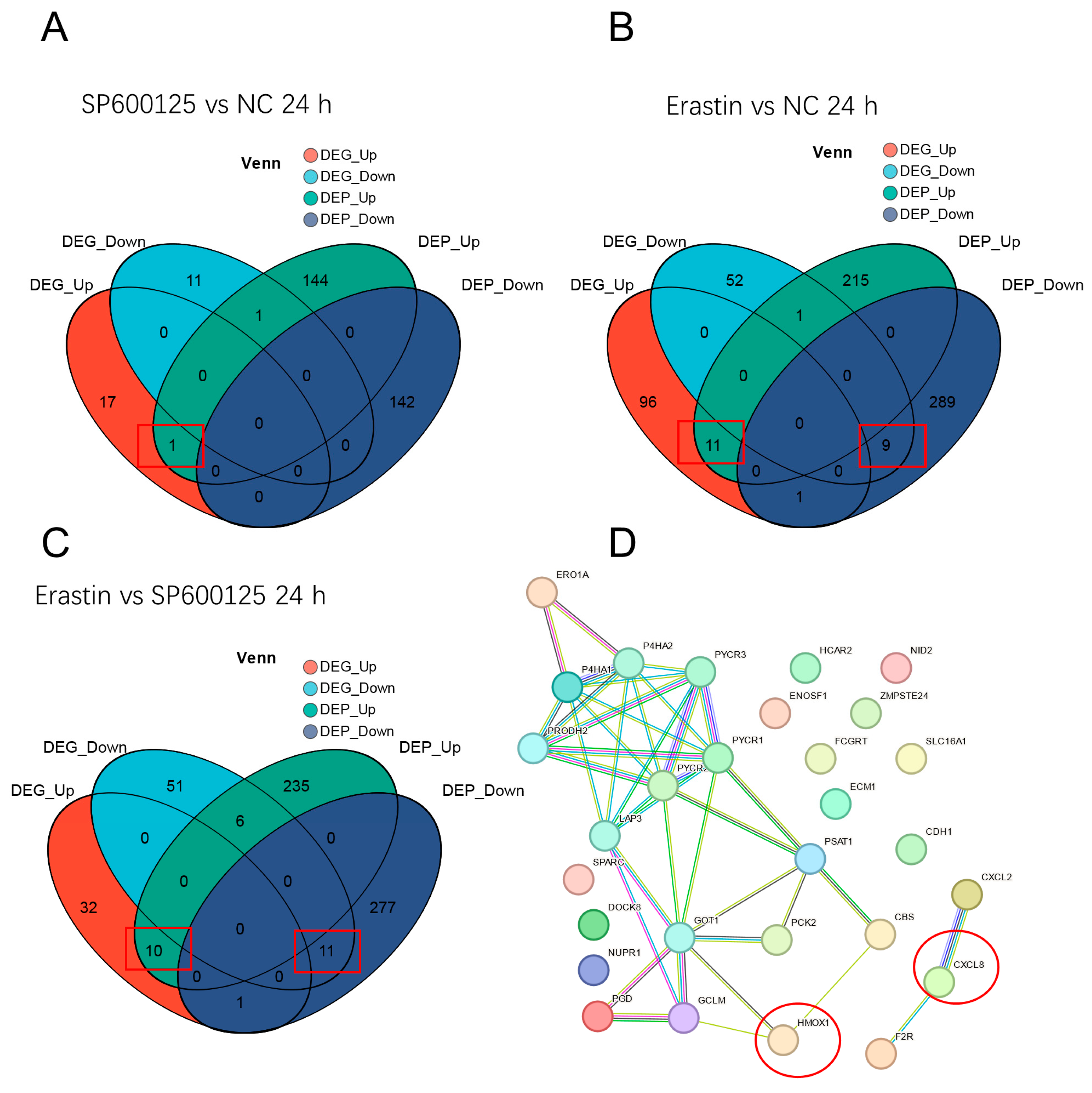
3.7. Conjoint Analysis of DEGs and DEPs and Protein–Protein Interaction Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chai, C.; Wang, Q.; Cao, S.; Zhao, Q.; Wen, Y.; Huang, X.; Wen, X.; Yan, Q.; Ma, X.; Wu, R. Serological and molecular epidemiology of Japanese encephalitis virus infections in swine herds in China, 2006–2012. J. Vet. Sci. 2018, 19, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.M.; Luo, J.; Chen, L.; Teng, M.; Bu, D.; Wang, F.Y.; Wang, L.; Wang, C.Q.; Zhang, G.P. Genetic analysis of strains of Japanese Encephalitis Virus isolated from swine in central China. Virus Genes 2010, 40, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jing, J.; Li, X.; Wang, J.; Feng, X.; Cao, R.; Chen, P. Integration analysis of miRNA and mRNA expression profiles in swine testis cells infected with Japanese encephalitis virus. Infect. Genet. Evol. 2015, 32, 342–347. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Xie, S.; Deng, W. Ferroptosis mechanisms and regulations in cardiovascular diseases in the past, present, and future. Cell Biol. Toxicol. 2024, 40, 17. [Google Scholar] [CrossRef]
- Jin, X.; Tang, J.; Qiu, X.; Nie, X.; Ou, S.; Wu, G.; Zhang, R.; Zhu, J. Ferroptosis: Emerging mechanisms, biological function, and therapeutic potential in cancer and inflammation. Cell Death Discov. 2024, 10, 45. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience. Cancer Lett. 2023, 559, 216119. [Google Scholar] [CrossRef]
- Chen, J.; Fu, J.; Zhao, S.; Zhang, X.; Chao, Y.; Pan, Q.; Sun, H.; Zhang, J.; Li, B.; Xue, T.; et al. Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis. Vet. Sci. 2023, 10, 456. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J.F. Ferroptosis: A mixed blessing for infectious diseases. Front. Pharmacol. 2022, 13, 992734. [Google Scholar] [CrossRef]
- Huang, R.; Wu, J.; Ma, Y.; Kang, K. Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis. Viruses 2023, 15, 2373. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.P.; Joshua, B.; Jin, N.Y.; Du, S.W.; Li, C. Ferroptosis in viral infection: The unexplored possibility. Acta Pharmacol. Sin. 2022, 43, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Tao, J.; Li, B.; Shi, Y.; Liu, H. Swine influenza virus triggers ferroptosis in A549 cells to enhance virus replication. Virol. J. 2022, 19, 104. [Google Scholar] [CrossRef] [PubMed]
- Anandhan, A.; Dodson, M.; Shakya, A.; Chen, J.J.; Liu, P.F.; Wei, Y.Y.; Tan, H.; Wang, Q.; Jiang, Z.Y.; Yang, K.; et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci. Adv. 2023, 9, ade9585. [Google Scholar] [CrossRef]
- Luo, L.X.; Zhang, Z.T.; Weng, Y.M.; Zeng, J.Y. Ferroptosis-Related Gene GCLC is a Novel Prognostic Molecular and Correlates with Immune Infiltrates in Lung Adenocarcinoma. Cells 2022, 11, 3371. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Zhu, S.X.; Li, F.Y. Systematical analysis of ferroptosis regulators and identification of GCLM as a tumor promotor and immunological biomarker in bladder cancer. Front. Oncol. 2022, 12, 1040892. [Google Scholar] [CrossRef]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.; Bao, H.; Tang, C.; Dong, X.; Yang, Q.; Yan, Y.; Yin, Z.; et al. The mechanism of ferroptosis and its related diseases. Mol. Biomed. 2023, 4, 33. [Google Scholar] [CrossRef]
- Dar, N.J.; John, U.; Bano, N.; Khan, S.; Bhat, S.A. Oxytosis/Ferroptosis in Neurodegeneration: The Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol. Neurobiol. 2024, 61, 1507–1526. [Google Scholar] [CrossRef]
- Xu, S.J.; Wu, B.X.; Zhong, B.Y.; Lin, L.Q.; Ding, Y.N.; Jin, X.; Huang, Z.W.; Lin, M.Y.; Wu, H.L.; Xu, D.P. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2)/System xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered 2021, 12, 10924–10934. [Google Scholar] [CrossRef]
- Xiang, Y.; Song, X.H.; Long, D.X. Ferroptosis regulation through Nrf2 and implications for neurodegenerative diseases. Arch. Toxicol. 2024, 98, 579–615. [Google Scholar] [CrossRef]
- Tian, J.; Fu, W.; Xie, Z.; Zhao, Y.; Yang, H.; Zhao, J. Methionine enkephalin (MENK) protected macrophages from ferroptosis by downregulating HMOX1 and ferritin. Proteome Sci. 2024, 22, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.X.; Duan, K.L.; Huang, Z.X.; Xue, Z.A.; Liang, J.Y.; Dang, Y.J.; Zhang, A.; Xiong, Y.; Ding, C.Y.; Guan, K.L.; et al. Tanshinone functions as a coenzyme that confers gain of function of NQO1 to suppress ferroptosis. Life Sci. Alliance 2022, 6, e202201667. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Xu, F.F.; Shen, J.C.; Xu, S.H. Identification of a Ferroptosis-Related Prognostic Gene PTGS2 Based on Risk Modeling and Immune Microenvironment of Early-Stage Cervical Cancer. J. Oncol. 2022, 2022, 3997562. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.L.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef]
- Hiebert, P. The Nrf2 transcription factor: A multifaceted regulator of the extracellular matrix. Matrix Biol. Plus 2021, 10, 100057. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer Cell 2024, 42, 513–534. [Google Scholar] [CrossRef]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, J.; Ren, S.; Zhang, Z.; Niu, K.; Li, H.; Wu, W.; Peng, C. The role of ferroptosis in virus infections. Front. Microbiol. 2023, 14, 1279655. [Google Scholar] [CrossRef]
- Zhou, R.; Wei, K.X.; Li, X.Y.; Yan, B.B.; Li, L. Mechanisms of ferroptosis and the relationship between ferroptosis and ER stress after JEV and HSV infection. Front. Microbiol. 2024, 15, 1415417. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Ying, M.; Jin, C.; Li, J.; Hu, X. Lactate dehydrogenases amplify reactive oxygen species in cancer cells in response to oxidative stimuli. Signal Transduct. Target. Ther. 2021, 6, 242. [Google Scholar] [CrossRef] [PubMed]
- Mokwatsi, G.G.; Schutte, A.E.; Kruger, R. A biomarker of tissue damage, lactate dehydrogenase, is associated with fibulin-1 and oxidative stress in blacks: The SAfrEIC study. Biomarkers 2016, 21, 48–55. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Tang, D.L.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Shin, D.; Kim, E.H.; Lee, J.; Roh, J.L. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic. Biol. Med. 2018, 129, 454–462. [Google Scholar] [CrossRef]
- Romani, P.; Nirchio, N.; Arboit, M.; Barbieri, V.; Tosi, A.; Michielin, F.; Shibuya, S.; Benoist, T.; Wu, D.C.; Hindmarch, C.C.T.; et al. Mitochondrial fission links ECM mechanotransduction to metabolic redox homeostasis and metastatic chemotherapy resistance. Nat. Cell Biol. 2022, 24, 168–180. [Google Scholar] [CrossRef]
- Li, Y.J.; Zeng, X.L.; Lu, D.H.; Yin, M.N.; Shan, M.R.; Gao, Y. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis. Hum. Reprod. 2021, 36, 951–964. [Google Scholar] [CrossRef]
- Huo, H.Z.; Zhou, Z.Y.; Qin, J.; Liu, W.Y.; Wang, B.; Gu, Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS ONE 2016, 11, e0154605. [Google Scholar] [CrossRef]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Vaishnav, D.; Jambal, P.; Reusch, J.E.; Pugazhenthi, S. SP600125, an inhibitor of c-jun N-terminal kinase, activates CREB by a p38 MAPK-mediated pathway. Biochem. Biophys. Res. Commun. 2003, 307, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Xing, J.; Liang, J.; Liu, S.; Huang, L.; Hu, P.; Liu, L.; Liao, M.; Qi, W. Japanese encephalitis virus restricts HMGB1 expression to maintain MAPK pathway activation for viral replication. Vet. Microbiol. 2021, 262, 109237. [Google Scholar] [CrossRef]
- Mancinelli, R.; Rosa, L.; Cutone, A.; Lepanto, M.S.; Franchitto, A.; Onori, P.; Gaudio, E.; Valenti, P. Viral Hepatitis and Iron Dysregulation: Molecular Pathways and the Role of Lactoferrin. Molecules 2020, 25, 1997. [Google Scholar] [CrossRef]
- Vogt, A.C.S.; Arsiwala, T.; Mohsen, M.; Vogel, M.; Manolova, V.; Bachmann, M.F. On Iron Metabolism and Its Regulation. Int. J. Mol. Sci. 2021, 22, 4591. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, Z.M.; Yi, X.; Wei, X.; Jiang, D.S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef]
- Li, J.Y.; Yao, Y.M.; Tian, Y.P. Ferroptosis: A Trigger of Proinflammatory State Progression to Immunogenicity in Necroinflammatory Disease. Front. Immunol. 2021, 12, 701163. [Google Scholar] [CrossRef]
- Chai, Z.; Ma, T.; Li, Y.; Chen, Q.; Kang, Y.; Sun, J.; Peng, T.; Wang, N.; Yu, C.; Wang, L.; et al. Inhibition of inflammatory factor TNF-alpha by ferrostatin-1 in microglia regulates necroptosis of oligodendrocyte precursor cells. Neuroreport 2023, 34, 583–591. [Google Scholar] [CrossRef]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Garg, A.; Dhole, T.N.; Chaturvedi, R. Exaggerated levels of some specific TLRs, cytokines and chemokines in Japanese encephalitis infected BV2 and neuro 2A cell lines associated with worst outcome. Virol. J. 2023, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.Y.; Gao, Y.; Zhang, J.Q.; Zhang, H.; Xie, C.Z.; Nan, F.L.; Feng, S.; Ha, Z.; Li, C.H.; Zhu, X.Y.; et al. Toll-like receptor 2 signaling pathway activation contributes to a highly efficient inflammatory response in Japanese encephalitis virus-infected mouse microglial cells by proteomics. Front. Microbiol. 2022, 13, 989183. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Zuo, Z.; Di, X.; Huang, Y.; Gong, G.; Xu, B.; Wang, L.; Zhang, X.; Liang, Z.; Hou, Y.; et al. Salidroside attenuates HALI via IL-17A-mediated ferroptosis of alveolar epithelial cells by regulating Act1-TRAF6-p38 MAPK pathway. Cell Commun. Signal 2022, 20, 183. [Google Scholar] [CrossRef]
- Song, J.; Zhang, H.; Tong, Y.; Wang, Y.; Xiang, Q.; Dai, H.; Weng, C.; Wang, L.; Fan, J.; Shuai, Y.; et al. Molecular mechanism of interleukin-17A regulating airway epithelial cell ferroptosis based on allergic asthma airway inflammation. Redox Biol. 2023, 68, 102970. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhen, Z.D.; Fan, D.Y.; Wang, P.G.; An, J. Transcriptomic Analysis Suggests the M1 Polarization and Launch of Diverse Programmed Cell Death Pathways in Japanese Encephalitis Virus-Infected Macrophages. Viruses 2020, 12, 356. [Google Scholar] [CrossRef]
- He, J.P.; Abikoye, A.M.; McLaughlin, B.P.; Middleton, R.S.; Sheldon, R.; Jones, R.G.; Schafer, Z.T. Reprogramming of iron metabolism confers ferroptosis resistance in ECM-detached cells. iScience 2023, 26, 106827. [Google Scholar] [CrossRef]
- Esteves, A.D.; Koyuncu, O.O.; Enquist, L.W. A Pseudorabies Virus Serine/Threonine Kinase, US3, Promotes Retrograde Transport in Axons via Akt/mToRC1. J. Virol. 2022, 96, e0175221. [Google Scholar] [CrossRef]
- Ferrara, G.; Longobardi, C.; Damiano, S.; Ciarcia, R.; Pagnini, U.; Montagnaro, S. Modifications of the PI3K/Akt/mTOR axis during FeHV-1 infection in permissive cells. Front. Vet. Sci. 2023, 10, 1157350. [Google Scholar] [CrossRef]
- Zhang, G.; Mi, W.A.; Wang, C.Y.; Li, J.H.; Zhang, Y.Z.; Liu, N.N.; Jiang, M.M.; Jia, G.Y.; Wang, F.; Yang, G.; et al. Targeting AKT induced Ferroptosis through FTO/YTHDF2-dependent GPX4 m6A methylation up-regulating and degradating in colorectal cancer. Cell Death Discov. 2023, 9, 457. [Google Scholar] [CrossRef]
- Su, H.; Peng, C.; Liu, Y. Regulation of ferroptosis by PI3K/Akt signaling pathway: A promising therapeutic axis in cancer. Front. Cell Dev. Biol. 2024, 12, 1372330. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Lekshmi, V.S.; Asha, K.; Sanicas, M.; Asi, A.; Arya, U.M.; Kumar, B. PI3K/Akt/Nrf2 mediated cellular signaling and virus-host interactions: Latest updates on the potential therapeutic management of SARS-CoV-2 infection. Front. Mol. Biosci. 2023, 10, 1158133. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Y.; Wei, Y.; Liu, G.; Liu, Y.; Gao, Q.; Zou, L.; Zeng, W.; Zhang, N. Activation of AKT pathway by Nrf2/PDGFA feedback loop contributes to HCC progression. Oncotarget 2016, 7, 65389–65402. [Google Scholar] [CrossRef]
- Zhong, F.M.; Yao, F.Y.; Liu, J.; Zhang, H.B.; Zhang, J.; Zhang, N.; Lin, J.; Li, S.Q.; Li, M.Y.; Jiang, J.Y.; et al. Ferroptosis-related molecular patterns reveal immune escape, inflammatory development and lipid metabolism characteristics of the tumor microenvironment in acute myeloid leukemia. Front. Oncol. 2022, 12, 888570. [Google Scholar] [CrossRef]
- Kim, J.H.; Patil, A.M.; Choi, J.Y.; Kim, S.B.; Uyangaa, E.; Hossain, F.M.; Park, S.Y.; Lee, J.H.; Eo, S.K. CCR5 ameliorates Japanese encephalitis via dictating the equilibrium of regulatory CD4+Foxp3+ T and IL-17+CD4+ Th17 cells. J. Neuroinflamm. 2016, 13, 223. [Google Scholar] [CrossRef]
- Lannes, N.; Summerfield, A.; Filgueira, L. Regulation of inflammation in Japanese encephalitis. J. Neuroinflamm. 2017, 14, 158. [Google Scholar] [CrossRef]
- Das, S.; Dutta, K.; Kumawat, K.L.; Ghoshal, A.; Adhya, D.; Basu, A. Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese encephalitis. PLoS ONE 2011, 6, e17225. [Google Scholar] [CrossRef]
- Wu, D.Q.; Hu, Q.; Wang, Y.Q.; Jin, M.Y.; Tao, Z.Q.; Wan, J. Identification of HMOX1 as a Critical Ferroptosis-Related Gene in Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 833642. [Google Scholar] [CrossRef]
- Liao, S.J.; Huang, M.; Liao, Y.L.; Yuan, C. HMOX1 Promotes Ferroptosis Induced by Erastin in Lens Epithelial Cell through Modulates Fe Production. Curr. Eye Res. 2023, 48, 25–33. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Bai, X.; Lin, T.; Wang, X.Y.; Zhang, B.H.; Dai, L.J.; Shi, J.; Zhang, Y.; Zhao, X.X. HMOX1 Promotes Ferroptosis in Mammary Epithelial Cells via FTH1 and Is Involved in the Development of Clinical Mastitis in Dairy Cows. Antioxidants 2022, 11, 2221. [Google Scholar] [CrossRef] [PubMed]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System Xc−/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Matsumoto, M.; Shindo, T.; Saigusa, D.; Kato, H.; Suzuki, K.; Sato, M.; Ishii, Y.; Shimokawa, H.; Igarashi, K. Ferroptosis is controlled by the coordinated transcriptional regulation of glutathione and labile iron metabolism by the transcription factor BACH1. J. Biol. Chem. 2020, 295, 69–82. [Google Scholar] [CrossRef]
- Liu, J.J.; Lu, X.Y.; Zeng, S.Y.; Fu, R.; Wang, X.D.; Luo, L.T.; Huang, T.; Deng, X.S.; Zheng, H.L.; Ma, S.Q.; et al. ATF3-CBS signaling axis coordinates ferroptosis and tumorigenesis in colorectal cancer. Redox Biol. 2024, 71, 103118. [Google Scholar] [CrossRef]
- Liu, N.; Lin, X.L.; Huang, C.Y. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292. [Google Scholar] [CrossRef]
- Wu, X.W.; Wang, S.N.; Wang, C.C.; Wu, C.W.; Zhao, Z.Y. Bioinformatics analysis identifies coagulation factor II receptor as a potential biomarker in stomach adenocarcinoma. Sci. Rep. 2024, 14, 2468. [Google Scholar] [CrossRef]
- Kraaij, M.D.; Savage, N.D.; van der Kooij, S.W.; Koekkoek, K.; Wang, J.; van den Berg, J.M.; Ottenhoff, T.H.; Kuijpers, T.W.; Holmdahl, R.; van Kooten, C.; et al. Induction of regulatory T cells by macrophages is dependent on production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2010, 107, 17686–17691. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Yang, R.; Xiao, L.; Dong, S.; Lin, J.; Liu, G.; Shan, H. Erastin inhibits porcine epidemic diarrhea virus replication in Vero cells. Front. Cell. Infect. Microbiol. 2023, 13, 1142173. [Google Scholar] [CrossRef]
- Hu, J.C.; Zhu, T.P.; Gui, Y.C.; Tan, Z.B.; Wei, R.Q.; Hu, B.L.; Xu, J.W. miR-28-5p inhibits carcinogenesis in colon cancer cells and is necessary for erastin-induced ferroptosis. Transl. Cancer Res. 2020, 9, 2931–2940. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Chen, Y.; Kang, X.; Zhao, A.; Yang, S. Transcriptome and Proteome Analyses Revealed Differences in JEV-Infected PK-15 Cells in Response to Ferroptosis Agonists and Antagonists. Animals 2024, 14, 3516. https://doi.org/10.3390/ani14233516
Zhou X, Chen Y, Kang X, Zhao A, Yang S. Transcriptome and Proteome Analyses Revealed Differences in JEV-Infected PK-15 Cells in Response to Ferroptosis Agonists and Antagonists. Animals. 2024; 14(23):3516. https://doi.org/10.3390/ani14233516
Chicago/Turabian StyleZhou, Xiaolong, Yiwei Chen, Xinyao Kang, Ayong Zhao, and Songbai Yang. 2024. "Transcriptome and Proteome Analyses Revealed Differences in JEV-Infected PK-15 Cells in Response to Ferroptosis Agonists and Antagonists" Animals 14, no. 23: 3516. https://doi.org/10.3390/ani14233516
APA StyleZhou, X., Chen, Y., Kang, X., Zhao, A., & Yang, S. (2024). Transcriptome and Proteome Analyses Revealed Differences in JEV-Infected PK-15 Cells in Response to Ferroptosis Agonists and Antagonists. Animals, 14(23), 3516. https://doi.org/10.3390/ani14233516