
Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry
 , , , ,
, , , ,
Simple Summary
Abstract
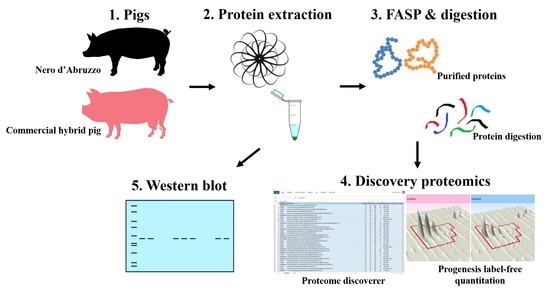
1. Introduction
2. Materials and Methods
2.1. Sampling and Sample Preparation for Label-Free LC-MS/MS Analysis
2.2. Mass Spectrometry for Label-Free LC/MS
2.3. Label-Free LC/MS Quantitative Profiling
2.4. Western Blot Analysis
2.5. Protein Classification
3. Results
3.1. Label-Free Proteomic Analysis of Pig Muscle Exudate
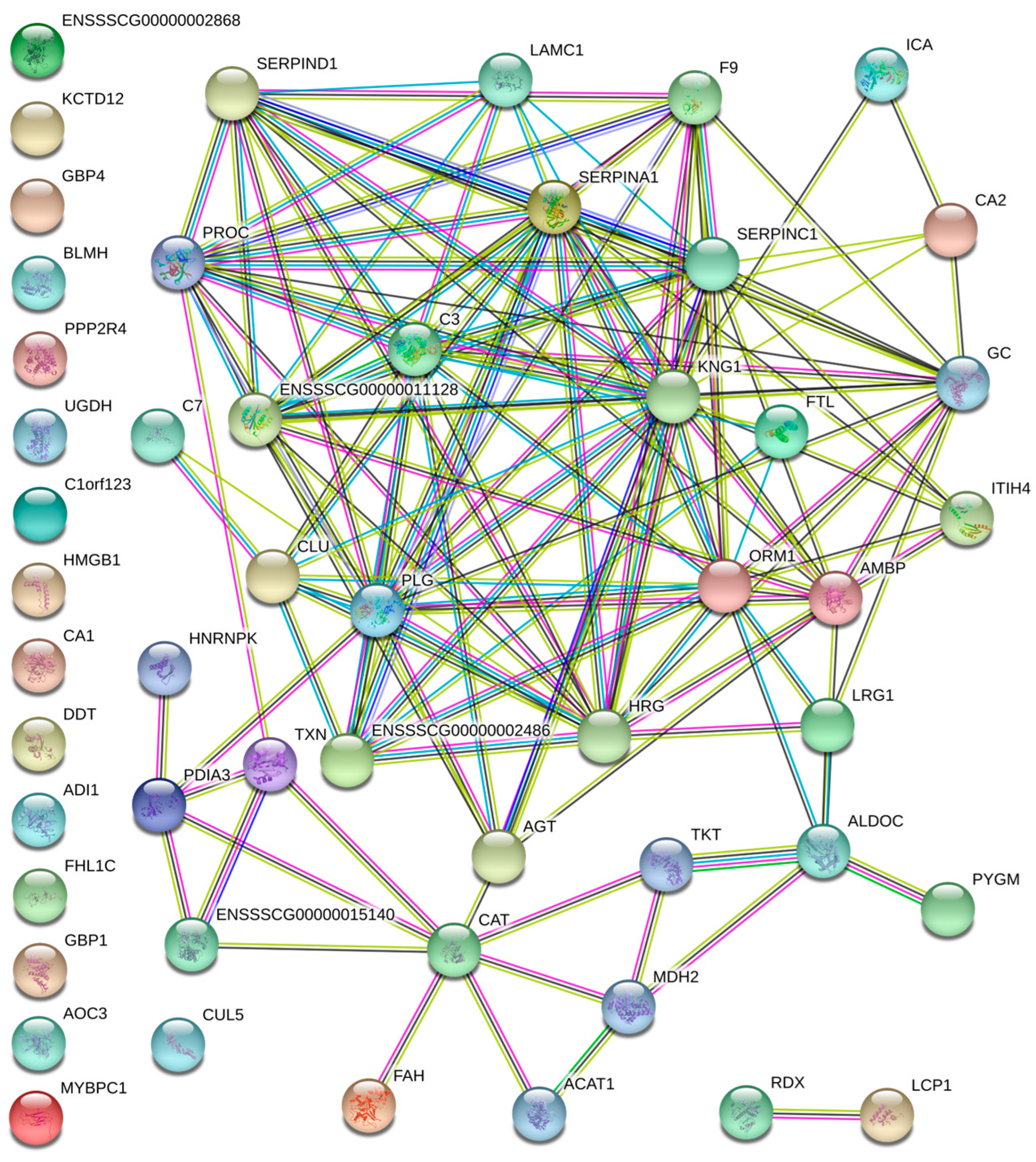
3.2. Protein–Protein Interaction Analysis
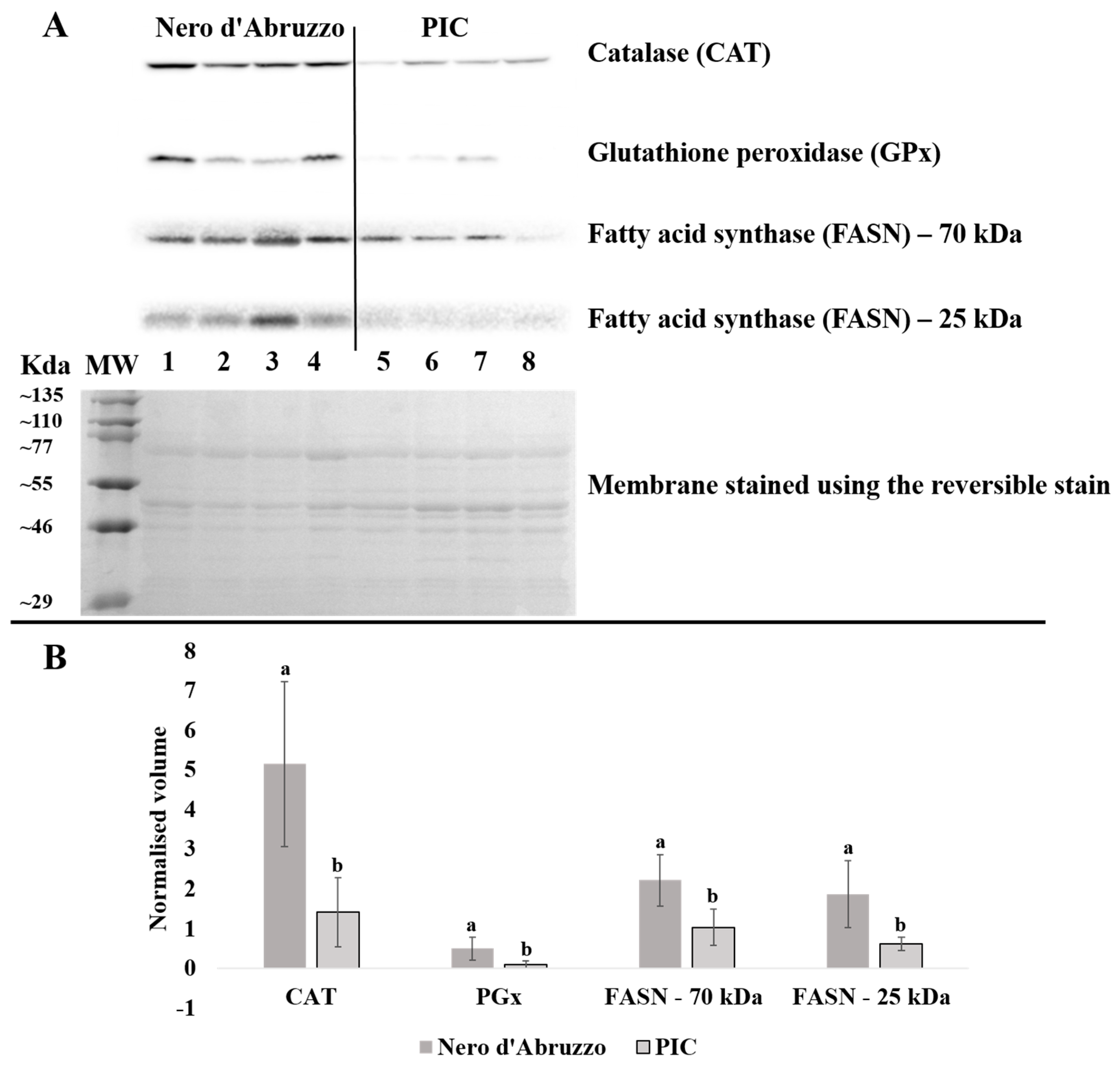
3.3. Confirmation of Differential Protein Expression by Western Blot
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olesen, I.; Groen, A.F.; Gjerde, B. Definition of animal breeding goals for sustainable production systems. J. Anim. Sci. 2000, 78, 570–582. [Google Scholar] [CrossRef]
- Mendelsohn, R. The challenge of conserving indigenous domesticated animals. Ecol. Econ. 2003, 45, 501–510. [Google Scholar] [CrossRef]
- Hiemstra, S.J.; de Haas, Y.; Mäkit-Tanila, A.; Gandini, G. Local Cattle Breeds in Europe: Development of Policies and Strategies for Self-Sustaining Breeds; Wageningen Academic Publishers: Wageningen, The Netherlands, 2010; ISBN 9789086861446. [Google Scholar]
- Lopez-Bote, C.J. Sustained utilization of the Iberian pig breed. Meat Sci. 1998, 49, S17–S27. [Google Scholar] [CrossRef]
- Edwards, S. Product quality attributes associated with outdoor pig production. Livest. Prod. Sci. 2005, 94, 5–14. [Google Scholar] [CrossRef]
- Verbeke, W.; Perez-Cueto, F.J.A.; de Barcellos, M.D.; Krystallis, A.; Grunert, K.G. European citizen and consumer attitudes and preferences regarding beef and pork. Meat Sci. 2010, 84, 284–292. [Google Scholar] [CrossRef]
- Jones, G. Genetic aspects of domestication, common breeds and their origin. In The Genetics of the Pig; Ruvinsky, A., Rothschild, M., Eds.; CAB International: Oxon, UK, 1998; pp. 17–50. [Google Scholar]
- Turner, J. Animal Breeding, Welfare and Society; Routledge: New York, NY, USA, 2010; pp. 1–324. [Google Scholar]
- Scherf, B.D. World Watch List for Domestic Animal Diversity; Fao, U., Ed.; Food and Agriculture Organization: Rome, Italy, 2000. [Google Scholar]
- Porter, V. Pigs: A Handbook to the Breeds of the World; Helm Information Ltd.: Robertsbridge, UK, 1993. [Google Scholar]
- Megens, H.-J.; Crooijmans, R.P.; San Cristobal, M.; Hui, X.; Li, N.; Groenen, M. Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: Differences in microsatellite variation between two areas of domestication. Genet. Sel. Evol. 2008, 40, 103–128. [Google Scholar] [CrossRef]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef]
- Murgiano, L.; D’alessandro, A.; Egidi, M.G.; Crisà, A.; Prosperini, G.; Timperio, A.M.; Valentini, A.; Zolla, L. Proteomics and Transcriptomics Investigation on longissimus Muscles in Large White and Casertana Pig Breeds. J. Proteome Res. 2010, 9, 6450–6466. [Google Scholar] [CrossRef]
- Bovo, S.; Di Luca, A.; Galimberti, G.; Dall’olio, S.; Fontanesi, L. A comparative analysis of label-free liquid chromatography-mass spectrometry liver proteomic profiles highlights metabolic differences between pig breeds. PLoS ONE 2018, 13, e0199649. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, A.; Mullen, A.M.; Elia, G.; Davey, G.; Hamill, R.M. Centrifugal drip is an accessible source for protein indicators of pork ageing and water-holding capacity. Meat Sci. 2011, 88, 261–270. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.; Henry, M.; Clynes, M.; Meleady, P. Filter-Aided Sample Preparation (FASP) for Improved Proteome Analysis of Recombinant Chinese Hamster Ovary Cells. Methods Mol. Biol. 2017, 1603, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, A.; Henry, M.; Meleady, P.; O’connor, R. Label-free LC-MS analysis of HER2+ breast cancer cell line response to HER2 inhibitor treatment. DARU J. Pharm. Sci. 2015, 23, 40. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Byrne, J.C.; Downes, M.R.; O’donoghue, N.; O’keane, C.; O’neill, A.; Fan, Y.; Fitzpatrick, J.M.; Dunn, M.J.; Watson, R.W.G. 2D-DIGE as a Strategy to Identify Serum Markers for the Progression of Prostate Cancer. J. Proteome Res. 2008, 8, 942–957. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Franci, O.; Pugliese, C. Italian autochthonous pigs: Progress report and research perspectives. Ital. J. Anim. Sci. 2007, 6, 663–671. [Google Scholar] [CrossRef]
- Bordoni, B.; Zanier, E. Anatomic connections of the diaphragm: Influence of respiration on the body system. J. Multidiscip. Healthc. 2013, 6, 281–291. [Google Scholar] [CrossRef]
- Di Luca, A.; Hamill, R.M.; Mullen, A.M.; Slavov, N.; Elia, G. Comparative Proteomic Profiling of Divergent Phenotypes for Water Holding Capacity across the Post Mortem Ageing Period in Porcine Muscle Exudate. PLoS ONE 2016, 11, e0150605. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, A.; Hamill, R.; Mullen, A.M.; Elia, G. Dige analysis of animal tissues. In Difference Gel Electrophoresis: Methods and Protocols; Humana Press: Totowa, NJ, USA, 2018; Volume 1664. [Google Scholar]
- Di Luca, A.; Ianni, A.; Bennato, F.; Henry, M.; Meleady, P.; Martino, G. A Label-Free Quantitative Analysis for the Search of Proteomic Differences between Goat Breeds. Animals 2022, 12, 3336. [Google Scholar] [CrossRef]
- Neilson, K.A.; Ali, N.A.; Muralidharan, S.; Mirzaei, M.; Mariani, M.; Assadourian, G.; Lee, A.; van Sluyter, S.C.; Haynes, P.A. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 2011, 11, 535–553. [Google Scholar] [CrossRef]
- Xu, Y.J.; Jin, M.L.; Wang, L.J.; Zhang, A.D.; Zuo, B.; Xu, D.Q.; Ren, Z.Q.; Lei, M.G.; Mo, X.Y.; Li, F.E.; et al. Differential proteome analysis of porcine skeletal muscles between Meishan and Large White. J. Anim. Sci. 2009, 87, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry 1989, 28, 4523–4530. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- McNeel, R.L.; Mersmann, H.J. Nutritional deprivation reduces the transcripts for transcription factors and adipocyte-characteristic proteins in porcine adipocytes. J. Nutr. Biochem. 2000, 11, 139–146. [Google Scholar] [CrossRef]
- Ding, S.-T.; Schinckel, A.P.; Weber, T.E.; Mersmann, H.J. Expression of porcine transcription factors and genes related to fatty acid metabolism in different tissues and genetic populations. J. Anim. Sci. 2000, 78, 2127–2134. [Google Scholar] [CrossRef]
- Mildner, A.M.; Clarke, S.D. Porcine Fatty Acid Synthase: Cloning of a Complementary DNA, Tissue Distribution of Its mRNA and Suppression of Expression by Somatotropin and Dietary Protein. J. Nutr. 1991, 121, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Cava, R.; Estevez, M.; Ruiz, J.; Morcuende, D. Physicochemical characteristics of three muscles from free-range reared Iberian pigs slaughtered at 90 kg live weight. Meat Sci. 2003, 63, 533–541. [Google Scholar] [CrossRef]
- Morales, J.; Pérez, J.; Baucells, M.; Mourot, J.; Gasa, J. Comparative digestibility and lipogenic activity in Landrace and Iberian finishing pigs fed ad libitum corn- and corn–sorghum–acorn-based diets. Livest. Prod. Sci. 2002, 77, 195–205. [Google Scholar] [CrossRef]
- Lorenzo, J.M.; Domínguez, R. Effect of genotype on fatty acid composition of intramuscular and subcutaneous fat of Celta pig breed. Grasas Aceites 2014, 65, e037. [Google Scholar] [CrossRef]
- Michiels, C.; Raes, M.; Toussaint, O.; Remacle, J. Importance of SE-glutathione peroxidase, catalase, and CU/ZN-SOD for cell survival against oxidative stress. Free Radic. Biol. Med. 1994, 17, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Martino, G.; Mugnai, C.; Compagnone, D.; Grotta, L.; Del Carlo, M.; Sarti, F. Comparison of Performance, Meat Lipids and Oxidative Status of Pigs from Commercial Breed and Organic Crossbreed. Animals 2014, 4, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.-B.; Chen, D.-W.; Zhang, K.-Y.; Yu, B. Effects of Oxidative Stress on Growth Performance, Nutrient Digestibilities and Activities of Antioxidative Enzymes of Weanling Pigs. Asian-Australas. J. Anim. Sci. 2007, 20, 1600–1605. [Google Scholar] [CrossRef]
- Frankič, T.; Salobir, J. In vivo antioxidant potential of Sweet chestnut (Castanea sativa Mill.) wood extract in young growing pigs exposed to n-3 PUFA-induced oxidative stress. J. Sci. Food Agric. 2011, 91, 1432–1439. [Google Scholar] [CrossRef] [PubMed]
- Jordán, M.J.; Martínez-Conesa, C.; Bañón, S.; Otal, J.; Quílez, M.; García-Aledo, I.; Romero-Espinar, P.; Sánchez-Gómez, P. The Combined Effect of Mediterranean Shrubland Pasture and the Dietary Administration of Sage By-Products on the Antioxidant Status of Segureña Ewes and Lambs. Antioxidants 2020, 9, 938. [Google Scholar] [CrossRef] [PubMed]
- Moreno, I.; Ladero, L.; Cava, R. Effect of the Iberian pig rearing system on blood plasma antioxidant status and oxidative stress biomarkers. Livest. Sci. 2020, 235, 104006. [Google Scholar] [CrossRef]
- Ponnampalam, E.N.; Kiani, A.; Santhiravel, S.; Holman, B.W.B.; Lauridsen, C.; Dunshea, F.R. The Importance of Dietary Antioxidants on Oxidative Stress, Meat and Milk Production, and Their Preservative Aspects in Farm Animals: Antioxidant Action, Animal Health, and Product Quality—Invited Review. Animals 2022, 12, 3279. [Google Scholar] [CrossRef]
- Selsby, J.T. Increased catalase expression improves muscle function in mdx mice. Exp. Physiol. 2011, 96, 194–202. [Google Scholar] [CrossRef]
- Sullivan-Gunn, M.J.; Lewandowski, P.A. Elevated hydrogen peroxide and decreased catalase and glutathione peroxidase protection are associated with aging sarcopenia. BMC Geriatr. 2013, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Fulle, S.; Protasi, F.; Di Tano, G.; Pietrangelo, T.; Beltramin, A.; Boncompagni, S.; Vecchiet, L.; Fanò, G. The contribution of reactive oxygen species to sarcopenia and muscle ageing. Exp. Gerontol. 2004, 39, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.L.; Gagnon, B.J.; Senf, S.M.; Hain, B.A.; Judge, A.R. Ros-mediated activation of NF-kappaB and Foxo during muscle disuse. Muscle Nerve 2010, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A.; Crowley, R.S.; Carlson, E.J.; Epstein, C.J.; Mohapatra, P.K. Overexpression of copper/zinc superoxide dismutase: A novel cause of murine muscular dystrophy. Ann. Neurol. 1998, 44, 381–386. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt (*) | Identification | Peptides § | Score ¥ | Anova (p) | Fold Change | Highest Condition ǂ |
|---|---|---|---|---|---|---|
| A0A287ARC8 | Serine/threonine-protein phosphatase 2A activator | 2 | 7.38 | 0.020 | 2.36 | NA |
| A0A287AYR8 | LIM domain binding 3 | 2 | 5.92 | 0.032 | 2.32 | NA |
| A0A287BPJ1 | Rab GDP dissociation inhibitor | 2 | 5.17 | 0.002 | 2.01 | NA |
| I3LP41 | Malate dehydrogenase | 2 | 5.23 | 0.022 | 2.71 | NA |
| A0A287BPN0 | Microtubule-associated protein | 2 | 6.77 | 0.046 | 1.7 | NA |
| F1SN68 | Alpha-1-acid glycoprotein | 2 | 6.39 | 0.004 | 3.96 | NA |
| F8SUW1 | Ubiquitin carboxyl-terminal hydrolase (Fragment) | 2 | 4.68 | 0.008 | 4.55 | NA |
| A0A287AI92 | Carbonic anhydrase 1 | 2 | 9.11 | 0.0002 | 3.81 | NA |
| F1S765 | Chromosome 1 open reading frame 123 | 2 | 4.87 | 0.0004 | 2.55 | NA |
| H6TBN0 | Thioredoxin | 2 | 5.83 | 0.009 | 4.95 | NA |
| A0A287BB72 | Serine/threonine-protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform | 2 | 5.90 | 0.003 | 2.96 | NA |
| F1SV14 | Radixin | 3 | 12.14 | 0.019 | 2.89 | NA |
| I3LIM2 | UDP-glucose 6-dehydrogenase | 3 | 9.82 | 0.008 | 3.05 | NA |
| F1RIP3 | Ferritin | 3 | 7.28 | 0.012 | 11.16 | NA |
| A0A287A363 | Afamin | 3 | 10.12 | 0.012 | 2.08 | NA |
| E0D7H7 | Leukotriene A(4) hydrolase | 3 | 9.16 | 0.009 | 2.57 | NA |
| I3LQS0 | Heterogeneous nuclear ribonucleoprotein K | 3 | 10.99 | 0.024 | 1.86 | NA |
| A0A287B9B3 | Serpin family F member 2 | 2 | 6.40 | 0.002 | 2.43 | NA |
| F1S1G8 | Amine oxidase | 2 | 5.50 | 0.022 | 2.54 | NA |
| F1SCF0 | Alpha-1-antitrypsin | 2 | 5.47 | 0.009 | 2.05 | NA |
| F1RQ75 | Coagulation factor IX | 3 | 7.81 | 0.007 | 5.52 | NA |
| F2Z594 | High mobility group protein B1 | 3 | 11.89 | 0.014 | 3.74 | NA |
| A0A286ZTU7 | SERPIN domain-containing protein | 2 | 9.51 | 0.037 | 26.56 | NA |
| A0A287AC34 | Talin 1 | 2 | 5.72 | 0.0001 | 1.88 | NA |
| A0A286ZWJ9 | Succinate--CoA ligase [GDP-forming] subunit beta, mitochondrial | 2 | 4.93 | 0.007 | 3.17 | NA |
| L8AXL9 | IgG heavy chain | 2 | 5.09 | 0.035 | 72.24 | NA |
| A8D737 | Cadherin-13 | 2 | 7.12 | 0.018 | 1.84 | NA |
| A0A2C9F3B1 | Aldehyde dehydrogenase, mitochondrial | 2 | 6.35 | 0.0005 | 3 | NA |
| A0A287BGY0 | Actin, gamma 2, smooth muscle, enteric | 2 | 6.46 | 0.0001 | 17.47 | NA |
| F1SMJ1 | Complement component C7 | 2 | 5.67 | 0.008 | 1.66 | NA |
| E1CAJ5 | Protein disulfide-isomerase | 2 | 6.15 | 0.008 | 4.1 | NA |
| I3LP02 | Acetyl-CoA acetyltransferase 1 | 2 | 6.91 | 0.009 | 11.09 | NA |
| A0A287AFK2 | Troponin T, fast skeletal muscle | 2 | 6.38 | 0.005 | 4.14 | NA |
| A0A287A7R5 | IQ motif containing GTPase activating protein 1 | 2 | 8.21 | 0.003 | 1.98 | NA |
| A0A287A278 | Glutathione S-transferase theta-1 | 2 | 4.92 | 0.005 | 2.75 | NA |
| A0A286ZVF6 | GB1/RHD3-type G domain-containing protein | 2 | 8.02 | 0.036 | 5.99 | NA |
| F1RUM4 | Inter-alpha-trypsin inhibitor heavy chain H2 | 2 | 5.79 | 0.006 | 4.23 | NA |
| I3LRJ4 | Vitamin K-dependent protein C | 2 | 7.27 | 0.000 | 3.73 | NA |
| A0A5G2QYD6 | Cystatin domain-containing protein | 2 | 5.71 | 0.027 | 2.89 | NA |
| F1RHE6 | Potassium channel tetramerization domain containing 12 | 2 | 7.77 | 0.010 | 3.02 | NA |
| G8G223 | Coronin | 2 | 5.88 | 0.007 | 3.93 | NA |
| A0A287B8Z2 | Fructose-bisphosphate aldolase | 2 | 4.14 | 0.011 | 3.94 | NA |
| F1SN71 | Protein AMBP | 2 | 6.81 | 0.001 | 3.66 | NA |
| A0A287ARJ8 | SERPIN domain-containing protein | 2 | 4.89 | 0.010 | 14.51 | NA |
| I3LEI8 | 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase | 2 | 5.26 | 0.001 | 3.22 | NA |
| A0A287BIL8 | Endoplasmic reticulum chaperone BiP | 5 | 15.10 | 0.0005 | 2.99 | NA |
| L8B0S2 | IgG heavy chain | 4 | 12.08 | 0.009 | 2.92 | NA |
| A0A287AUT0 | GB1/RHD3-type G domain-containing protein | 5 | 19.67 | 0.011 | 9.82 | NA |
| F1S7K2 | Leucine rich alpha-2-glycoprotein 1 | 5 | 15.33 | 0.0001 | 14.13 | NA |
| F1RG45 | Angiotensinogen | 5 | 15.03 | 0.001 | 2.94 | NA |
| F1SBS4 | Complement C3 | 4 | 10.42 | 0.006 | 3.24 | NA |
| A0A068F143 | Glutathione peroxidase | 4 | 12.57 | 0.011 | 5.31 | NA |
| F1SRI8 | Myosin binding protein C, slow type | 4 | 12.05 | 0.009 | 2.33 | NA |
| A5A8W8 | Complement component 4A | 11 | 34.70 | 0.001 | 3.18 | NA |
| A0A287ARR1 | Ferritin | 4 | 10.50 | 0.001 | 23.26 | NA |
| F1RNW4 | Peptidase D | 5 | 14.54 | 0.001 | 2.34 | NA |
| Q58G70 | Fatty acid synthase (Fragment) | 9 | 25.74 | 0.033 | 16.62 | NA |
| A0A287BN06 | Pregnancy zone protein | 8 | 35.03 | 0.006 | 3.42 | NA |
| A0A287ANV7 | Lymphocyte cytosolic protein 1 | 9 | 29.80 | 0.006 | 3.86 | NA |
| F1SH92 | Inter-alpha-trypsin inhibitor heavy chain H4 | 10 | 31.34 | 0.012 | 4.4 | NA |
| I3LBF1 | Serotransferrin | 9 | 33.48 | 0.0003 | 3.1 | NA |
| A0A287A608 | AHNAK nucleoprotein | 6 | 14.82 | 0.001 | 4.18 | NA |
| B3STX9 | Prothrombin | 5 | 19.39 | 0.0001 | 3.8 | NA |
| F1SB81 | Plasminogen | 6 | 15.56 | 0.012 | 5.72 | NA |
| A0A287ADI8 | Alpha-1B-glycoprotein | 7 | 21.35 | 0.007 | 4.26 | NA |
| F2Z5E2 | SERPIN domain-containing protein | 6 | 17.77 | 0.001 | 1.92 | NA |
| A0A287AT21 | Sorbitol dehydrogenase | 4 | 25.71 | 0.004 | 3.07 | NA |
| F1RKY2 | Serpin family D member 1 | 3 | 8.88 | 0.003 | 2.52 | NA |
| F1SCC7 | SERPIN domain-containing protein | 3 | 7.22 | 0.019 | 5.02 | NA |
| I3L7D6 | Aldo_ket_red domain-containing protein | 3 | 7.50 | 0.025 | 2.9 | NA |
| A0A286ZIF4 | Glutathione S-transferase mu 4 | 3 | 10.38 | 0.013 | 2.26 | NA |
| A0A287BAY9 | Serum albumin | 3 | 8.92 | 0.002 | 3.32 | NA |
| I3LN42 | GC, vitamin D binding protein | 3 | 9.77 | 0.010 | 3.61 | NA |
| A0A287A3T6 | Ig-like domain-containing protein | 3 | 12.14 | 0.002 | 4.02 | NA |
| F1SFI4 | Kininogen 1 | 3 | 9.55 | 0.009 | 2.41 | NA |
| F1RIF3 | Fumarylacetoacetate hydrolase | 3 | 9.58 | 0.022 | 2.04 | NA |
| K7GSW5 | Clusterin | 4 | 10.98 | 0.003 | 4.02 | NA |
| K9IVR7 | WD repeat domain 1 | 3 | 7.95 | 0.001 | 2.45 | NA |
| A8U4R4 | Transketolase | 4 | 11.02 | 0.001 | 2.85 | NA |
| A0A287B7P2 | Leukocyte elastase inhibitor | 4 | 12.70 | 0.003 | 2.27 | NA |
| A0A287BJF1 | Myosin light chain 1 | 3 | 10.07 | 0.017 | 3.4 | NA |
| F1SGS9 | Catalase | 4 | 14.16 | 0.004 | 4.06 | NA |
| F1RXC2 | Carbonic anhydrase 2 | 3 | 9.19 | 0.021 | 2.01 | NA |
| UniProt (*) | Identification | Peptides § | Score ¥ | Anova (p) | Fold Change | Highest Condition ǂ |
|---|---|---|---|---|---|---|
| A0A286ZJK1 | Complement factor H | 2 | 6.68 | 0.004 | 1.76 | PIC |
| A0A287B8G0 | Pyruvate kinase | 6 | 24.40 | 0.011 | 1.48 | PIC |
| F1S9Q3 | Heat shock cognate 71 kDa protein | 2 | 6.26 | 0.001 | 2.86 | PIC |
| A0A287AQK7 | Heat shock protein HSP 90-alpha | 2 | 7.97 | 0.022 | 4.27 | PIC |
| F1RN71 | Bleomycin hydrolase | 2 | 5.46 | 0.013 | 1.93 | PIC |
| Q8WMK2 | Muscle creatine kinase (Fragment) | 3 | 13.15 | 0.014 | 2.76 | PIC |
| G9F6X8 | Protein disulfide-isomerase | 4 | 13.96 | 0.009 | 1.38 | PIC |
| A0A287BG23 | Glyceraldehyde-3-phosphate dehydrogenase | 6 | 19.88 | 0.002 | 2.64 | PIC |
| I3VKE6 | Ceruloplasmin | 4 | 16.43 | 0.0001 | 2.33 | PIC |
| A0A287A1V5 | Fructose-bisphosphate aldolase | 4 | 12.07 | 0.010 | 3.58 | PIC |
| F1RQQ8 | Alpha-1,4 glucan phosphorylase | 4 | 17.63 | 0.016 | 2.04 | PIC |
| A0A287BB89 | Pyridoxamine 5’-phosphate oxidase | 2 | 9.69 | 0.001 | 4.58 | PIC |
| F1S663 | Laminin subunit gamma 1 | 3 | 11.69 | 0.014 | 2.69 | PIC |
| A0A287A1Z9 | Eukaryotic translation initiation factor 4 gamma 1 | 3 | 10.28 | 0.0001 | 4.02 | PIC |
| I3LFR2 | Cullin 5 | 2 | 5.02 | 0.013 | 2.83 | PIC |
| K7GSI9 | Four and a half LIM domains 1 | 2 | 6.02 | 0.034 | 1.82 | PIC |
| A0A287AKT7 | Protein arginine N-methyltransferase 5 | 2 | 8.02 | 0.007 | 2.18 | PIC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Luca, A.; Ianni, A.; Bennato, F.; Martino, C.; Henry, M.; Meleady, P.; Martino, G. Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry. Animals 2023, 13, 1460. https://doi.org/10.3390/ani13091460
Di Luca A, Ianni A, Bennato F, Martino C, Henry M, Meleady P, Martino G. Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry. Animals. 2023; 13(9):1460. https://doi.org/10.3390/ani13091460
Chicago/Turabian StyleDi Luca, Alessio, Andrea Ianni, Francesca Bennato, Camillo Martino, Michael Henry, Paula Meleady, and Giuseppe Martino. 2023. "Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry" Animals 13, no. 9: 1460. https://doi.org/10.3390/ani13091460
APA StyleDi Luca, A., Ianni, A., Bennato, F., Martino, C., Henry, M., Meleady, P., & Martino, G. (2023). Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry. Animals, 13(9), 1460. https://doi.org/10.3390/ani13091460