
Detecting Forest Musk Deer Abscess Disease Pathogens Using 16S rRNA High-Throughput Sequencing Technology

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
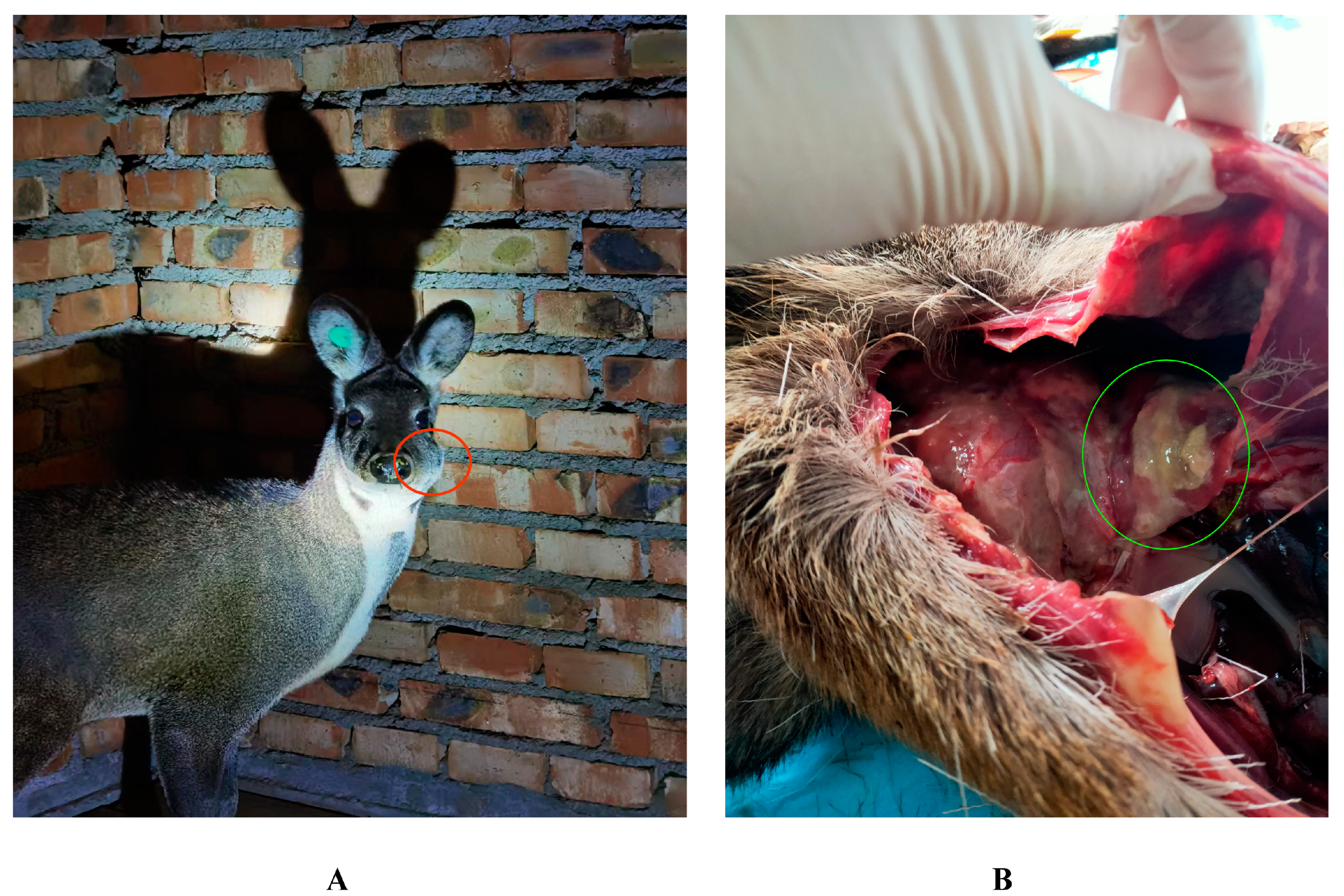
2.1. Pus Sample Collection
2.2. DNA Extraction and Amplification
2.3. 16S rRNA Gene Amplicon Sequencing and Data Processing
2.4. Statistical Analysis
3. Results
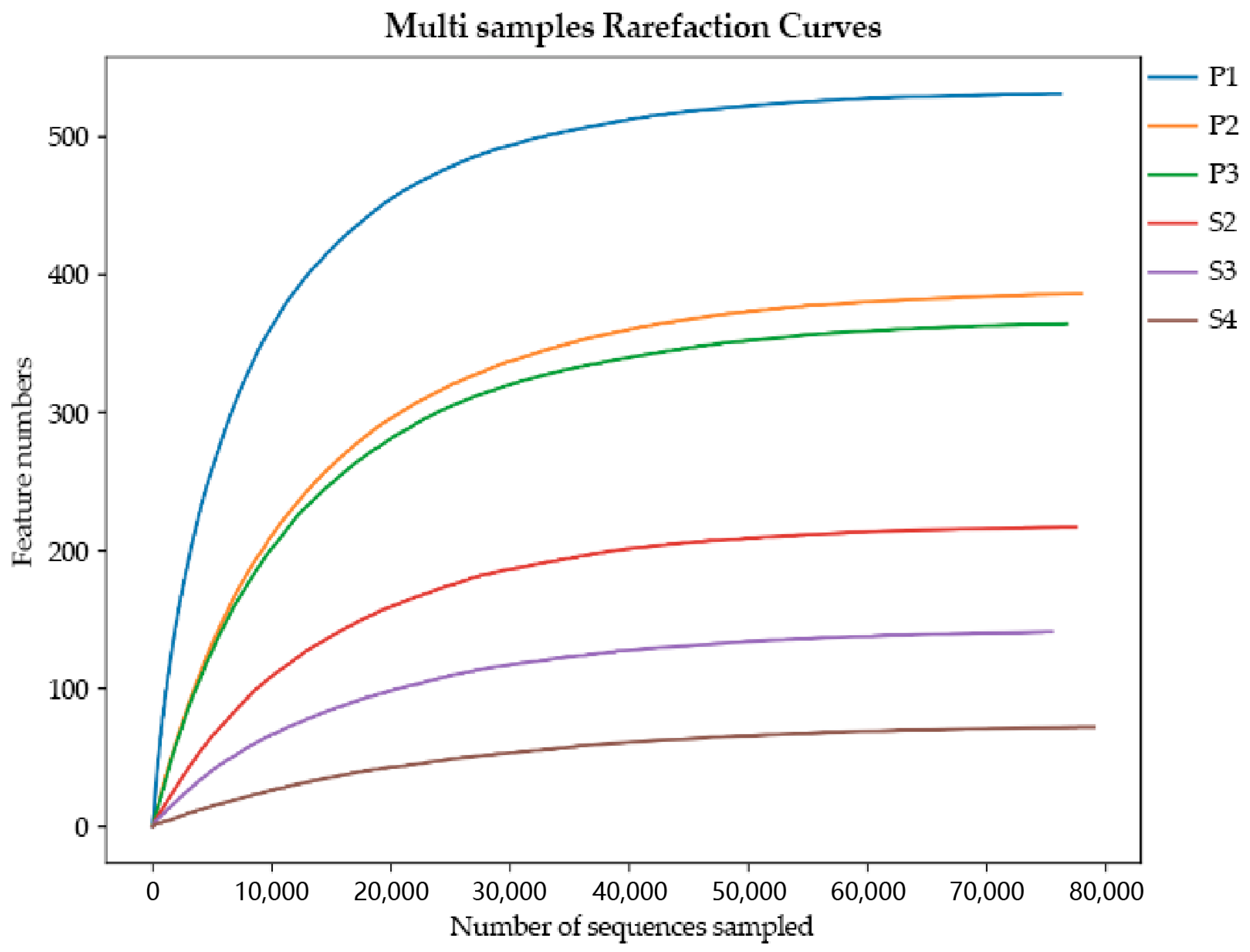
3.1. Sequence Statistics
3.2. Microbial Composition Analysis
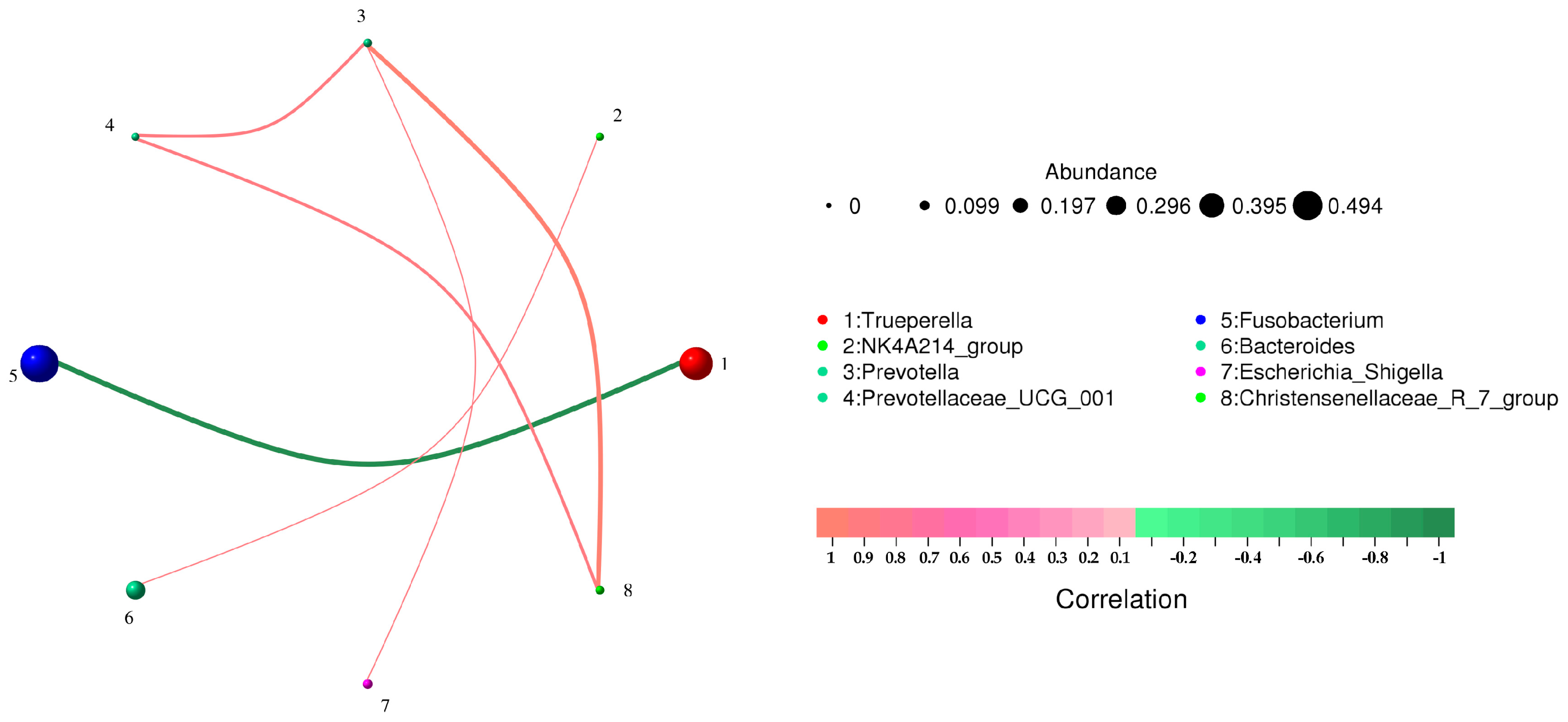
3.3. Correlation Analysis of Microbial Community Composition
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, Q.S.; Meng, X.X.; Xia, L.; Feng, Z.J. Conservation status and causes of decline of musk deer (Moschus spp.) in China. Biol. Conserv. 2003, 109, 333–342. [Google Scholar] [CrossRef]
- He, L.; Li, L.-H.; Wang, W.-X.; Liu, G.; Liu, S.-Q.; Liu, W.-H.; Hu, D.-F. Welfare of farmed musk deer: Changes in the biological characteristics of musk deer in farming environments. Appl. Anim. Behav. Sci. 2014, 156, 1–5. [Google Scholar]
- Chen, Q.; Zhao, K.; Li, H.; Liu, K.; Li, J.; Chu, Y.; Prithiviraj, B.; Yue, B.; Zhang, X. Antibacterial and anti-virulence effects of furazolidone on Trueperella pyogenes and Pseudomonas aeruginosa. BMC Vet. Res. 2022, 18, 114. [Google Scholar]
- Zhao, K.L.; Liu, Y.; Zhang, X.Y.; Palahati, P.; Wang, H.N.; Yue, B.S. Detection and characterization of antibiotic-resistance genes in Arcanobacterium pyogenes strains from abscesses of forest musk deer. J. Med. Microbiol. 2011, 60, 1820–1826. [Google Scholar]
- Luo, Y.; Cheng, J.G. Pathogen isolation and identification of Escherichia coli purulent disease of musk deer. Heilongjiang Anim. Sci. Vet. Med. 2006, 42, 81–83. (In Chinese) [Google Scholar]
- Sun, X. Study on the etiology of purulent disease of musk deer. Sichuan Anim. Vet. Sci. 2001, 28, 25–27. (In Chinese) [Google Scholar]
- Zhao, K.; Tian, Y.; Yue, B.; Wang, H.; Zhang, X. Virulence determinants and biofilm production among Trueperella pyogenes recovered from abscesses of captive forest musk deer. Arch. Microbiol. 2013, 195, 203–209. [Google Scholar]
- Wu, S.Z.; Yang, F.L. Isolation and Identification of Pathogenic Bacteria for Abscess in Captive Forest Musk Deer (Moschus berezovskii). Chin. J. Wildl. 2021, 42, 872–878. (In Chinese) [Google Scholar]
- Loeffelholz, M.; Fofanov, Y. The main challenges that remain in applying high-throughput sequencing to clinical diagnostics. Expert Rev. Mol. Diagn. 2015, 15, 1405–1408. [Google Scholar] [CrossRef][Green Version]
- Addis, M.F.; Tanca, A.; Uzzau, S.; Oikonomou, G.; Bicalho, R.C.; Moroni, P. The bovine milk microbiota: Insights and perspectives from-omics studies. Mol. Biosyst. 2016, 12, 2359–2372. [Google Scholar]
- Georgios, O.; Vinicius, S.M.; Carlos, S.; Ynte, H.S.; Rodrigo, C.B. Microbial diversity of bovine mastitic milk as described by pyrosequencing of metagenomic 16s rDNA. PLoS ONE 2017, 7, e47671. [Google Scholar]
- Oultram, J.W.H.; Ganda, E.K.; Boulding, S.C.; Bicalho, R.C.; Oikonomou, G. A Metataxonomic Approach Could Be Considered for Cattle Clinical Mastitis Diagnostics. Front. Vet. Sci. 2017, 4, 36. [Google Scholar] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar]
- Zhao, K. Isolation and Identification on Pathogens of Musk Deer Abscess Disease and Antibiotic Susceptibility Assay. Sichuan J. Zool. 2011, 30, 522–526. (In Chinese) [Google Scholar]
- Nagaraja, T.G.; Narayanan, S.K.; Stewart, G.C.; Chengappa, M.M. Fusobacterium necrophorum infections in animals: Pathogenesis and pathogenic mechanisms. Anaerobe 2005, 11, 239–246. [Google Scholar]
- Goto, T.; Tanaka, K.; Minh, T.C.; Watanabe, K. Complete sequence of pBFUK1, a carbapenemase-harboring mobilizable plasmid from Bacteroides fragilis, and distribution of pBFUK1-like plasmids among carbapenem-resistant B. fragilis clinical isolates. J. Antibiot. 2013, 66, 239–242. [Google Scholar]
- Jang, S.S.; Hirsh, D.C. Characterization, distribution, and microbiological associations of Fusobacterium spp. in clinical specimens of animal origin. J. Clin. Microbiol. 1994, 32, 384–387. [Google Scholar] [PubMed]
- Lechtenberg, K.F.; Nagaraja, T.G.; Leipold, H.W.; Chengappa, M.M. Bacteriologic and histologic studies of hepatic abscesses in cattle. Am. J. Vet. Res. 1988, 49, 58–62. [Google Scholar] [PubMed]
- Francis, A.M.; Jeon, S.J.; Cunha, F.; Jeong, K.C.; Galvao, K.N. Draft Genome Sequences of Two Fusobacterium necrophorum Strains Isolated from the Uterus of Dairy Cows with Metritis. Microbiol. Resour. Announc. 2019, 8, e00201-19. [Google Scholar] [PubMed]
- Ghimire, S.C.; Whittington, R.J.; Dhungyel, O.P.; Joshi, H.D.; Egerton, J.R. Diagnosis of footrot in goats: Application of ELISA tests for response to antigens of Dichelobacter nodosus. Vet. Microbiol. 2002, 87, 237–251. [Google Scholar] [PubMed]
- Shanthalingam, S.; Narayanan, S.; Batra, S.A.; Jegarubee, B.; Srikumaran, S. Fusobacterium necrophorum in North American Bighorn Sheep (Ovis canadensis) Pneumonia. J. Wildl. Dis. 2016, 52, 616–620. [Google Scholar]
- Brooks, J.W.; Kumar, A.; Narayanan, S.; Myers, S.; Brown, K.; Nagaraja, T.G.; Jayarao, B.M. Characterization of Fusobacterium isolates from the respiratory tract of white-tailed deer (Odocoileus virginianus). J. Vet. Diagn. Investig. 2014, 26, 213–220. [Google Scholar]
- Handeland, K.; Boye, M.; Bergsjo, B.; Bondal, H.; Isaksen, K.; Agerholm, J.S. Digital necrobacillosis in Norwegian wild tundra reindeer (Rangifer tarandus tarandus). J. Comp. Pathol. 2010, 143, 29–38. [Google Scholar]
- Kumar, A.; Anderson, D.; Amachawadi, R.G.; Nagaraja, T.G.; Narayanan, S.K. Characterization of Fusobacterium necrophorum isolated from llama and alpaca. J. Vet. Diagn. Investig. 2013, 25, 502–507. [Google Scholar]
- Chen, L.Z. Study on Isolation, Identification and Its Leukotoxin Immunological Characteristics of Bovine Origin Pathogenic Fusobacterium necrophorum. Ph.D. Thesis, Jilin University, Changchun, China, 2008. (In Chinese). [Google Scholar]
- Tan, Z.L.; Nagaraja, T.G.; Chengappa, M.M. Fusobacterium necrophorum infections: Virulence factors, pathogenic mechanism and control measures. Vet. Res. Commun. 1996, 20, 113–140. [Google Scholar]
- Tan, Z.L.; Nagaraja, T.G.; Chengappa, M.M.; Smith, J.S. Biological and biochemical characterization of Fusobacterium necrophorum leukotoxin. Am. J. Vet. Res. 1994, 55, 515–521. [Google Scholar]
- Narayanan, S.K.; Nagaraja, T.G.; Chengappa, M.M.; Stewart, G.C. Leukotoxins of gram-negative bacteria. Vet. Microbiol. 2002, 84, 337–356. [Google Scholar]
- Forrester, L.J.; Campbell, B.J.; Berg, J.N.; Barrett, J.T. Aggregation of platelets by Fusobacterium necrophorum. J. Clin. Microbiol. 1985, 22, 245–249. [Google Scholar] [PubMed]
- Amachawadi, R.G.; Nagaraja, T.G. Liver abscesses in cattle: A review of incidence in Holsteins and of bacteriology and vaccine approaches to control in feedlot cattle. J. Anim. Sci. 2016, 94, 1620–1632. [Google Scholar] [PubMed]
- Takeuchi, S.; Nakajima, Y.; Hashimoto, K. Pathogenic synergism of Fusobacterium necrophorum and other bacteria in formation of liver abscess in BALB/c mice. Nihon Juigaku Zasshi 1983, 45, 775–781. [Google Scholar] [PubMed]
- Mancuso, G.; Midiri, A.; Biondo, C.; Beninati, C.; Gambuzza, M.; Macri, D.; Bellantoni, A.; Weintraub, A.; Espevik, T.; Teti, G. Bacteroides fragilis-derived lipopolysaccharide produces cell activation and lethal toxicity via toll-like receptor 4. Infect. Immun. 2005, 73, 5620–5627. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lesion | Sex | Strain No |
|---|---|---|
| Lung | Male | P1 |
| Lung | Male | P2 |
| Lung | Male | P3 |
| Hoof | Male | S2 |
| Hoof | Male | S3 |
| Face | Female | S4 |
| Simple | Species and Abundance at the Generic Level |
|---|---|
| P1 | Trueperella (32.82%) Bacteroides (48.23%) |
| P2 | Trueperella (95.38%) |
| P3 | Trueperella (84.44%) |
| S2 | Fusobacterium (97.94%) |
| S3 | Fusobacterium (98.65%) |
| S4 | Fusobacterium (99.52%) |
| Simple | Species and Abundance at the Species Level |
|---|---|
| P1 | Trueperella pyogenes (32.82%) Bacteroides fragilis (48.21%) |
| P2 | Trueperella pyogenes (95.38%) |
| P3 | Trueperella pyogenes (84.44%) |
| S2 | Fusobacterium necrophorum (97.94%) |
| S3 | Fusobacterium necrophorum (98.65%) |
| S4 | Fusobacterium necrophorum (99.52%) |
| Pathogenic Bacteria. | P1 | P2 | P3 | S2 | S3 | S4 |
|---|---|---|---|---|---|---|
| Fusobacterium_necrophorum | 0 | 0 | 0 | 97.94% | 98.65% | 99.52% |
| Trueperella_pyogenes | 32.82% | 95.38% | 84.44% | 0.013% | 0 | 0 |
| Bacteroides_fragilis | 48.21% | 0 | 0 | 0 | 0 | 0 |
| From | To | Corr | p Value |
|---|---|---|---|
| Trueperella | Fusobacterium | −0.985610761 | 0.000309086 |
| NK4A214_group | Bacteroides | 0.828571429 | 0.041562682 |
| Prevotella | Escherichia_Shigella | 0.828571429 | 0.041562682 |
| Prevotella | Christensenellaceae_R_7_group | 0.942857143 | 0.004804665 |
| Prevotellaceae_UCG_001 | Christensenellaceae_R_7_group | 0.845154255 | 0.034109423 |
| Prevotellaceae_UCG_001 | Prevotella | 0.845154255 | 0.034109423 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, G.; Wang, Z.; Zhang, B.; Zhou, Z.; Hu, D.; Zhang, D. Detecting Forest Musk Deer Abscess Disease Pathogens Using 16S rRNA High-Throughput Sequencing Technology. Animals 2023, 13, 3142. https://doi.org/10.3390/ani13193142
Lu G, Wang Z, Zhang B, Zhou Z, Hu D, Zhang D. Detecting Forest Musk Deer Abscess Disease Pathogens Using 16S rRNA High-Throughput Sequencing Technology. Animals. 2023; 13(19):3142. https://doi.org/10.3390/ani13193142
Chicago/Turabian StyleLu, Guanjie, Zhe Wang, Baofeng Zhang, Zhichao Zhou, Defu Hu, and Dong Zhang. 2023. "Detecting Forest Musk Deer Abscess Disease Pathogens Using 16S rRNA High-Throughput Sequencing Technology" Animals 13, no. 19: 3142. https://doi.org/10.3390/ani13193142
APA StyleLu, G., Wang, Z., Zhang, B., Zhou, Z., Hu, D., & Zhang, D. (2023). Detecting Forest Musk Deer Abscess Disease Pathogens Using 16S rRNA High-Throughput Sequencing Technology. Animals, 13(19), 3142. https://doi.org/10.3390/ani13193142