

Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell RNA Sequencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Slaughtering Experiment
2.2. Single-Cell RNA Sequencing Library Preparation
2.3. Quality Control
2.4. Dimensionality Reduction and Cell Clustering
2.5. Inference and Analysis of Cell-Cell Communication among Granulosa Cell Subpopulations
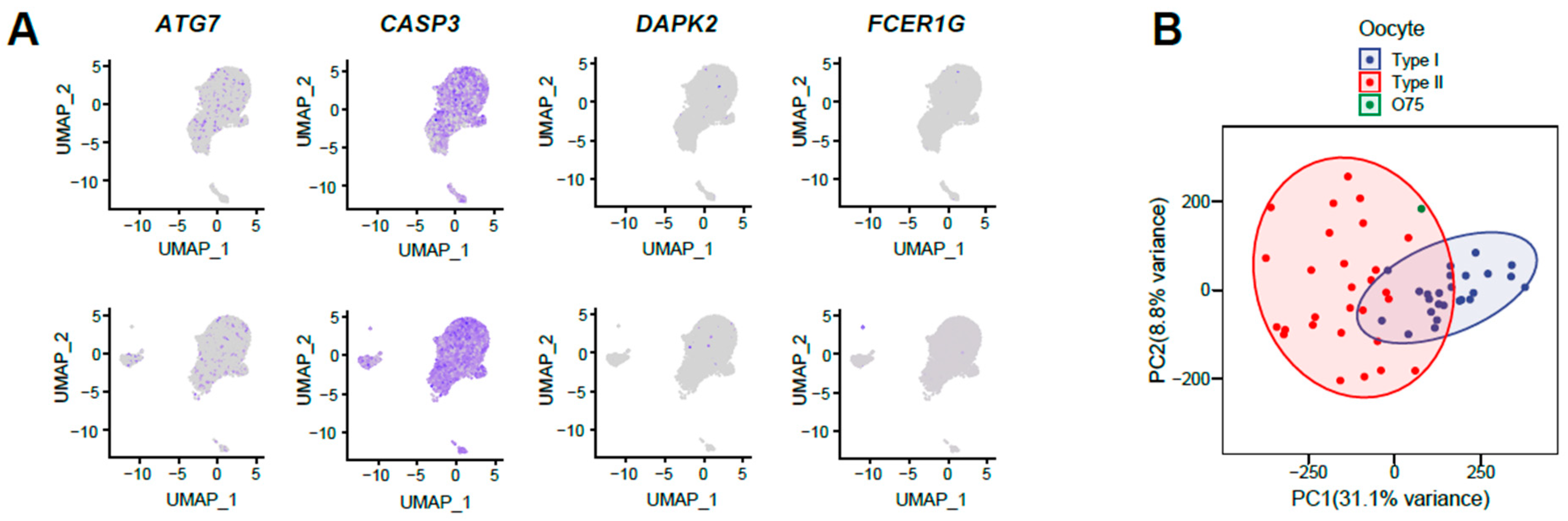
2.6. Single-Cell RNA-Seq Data Analysis of Oocytes
3. Results
3.1. Single-Cell Transcriptome Profiling Reveals Three Major Cell Types in Porcine Antral Follicles
3.2. Variations in Cell Distribution of Cumulus Granulosa Cells in Porcine Antral Follicles
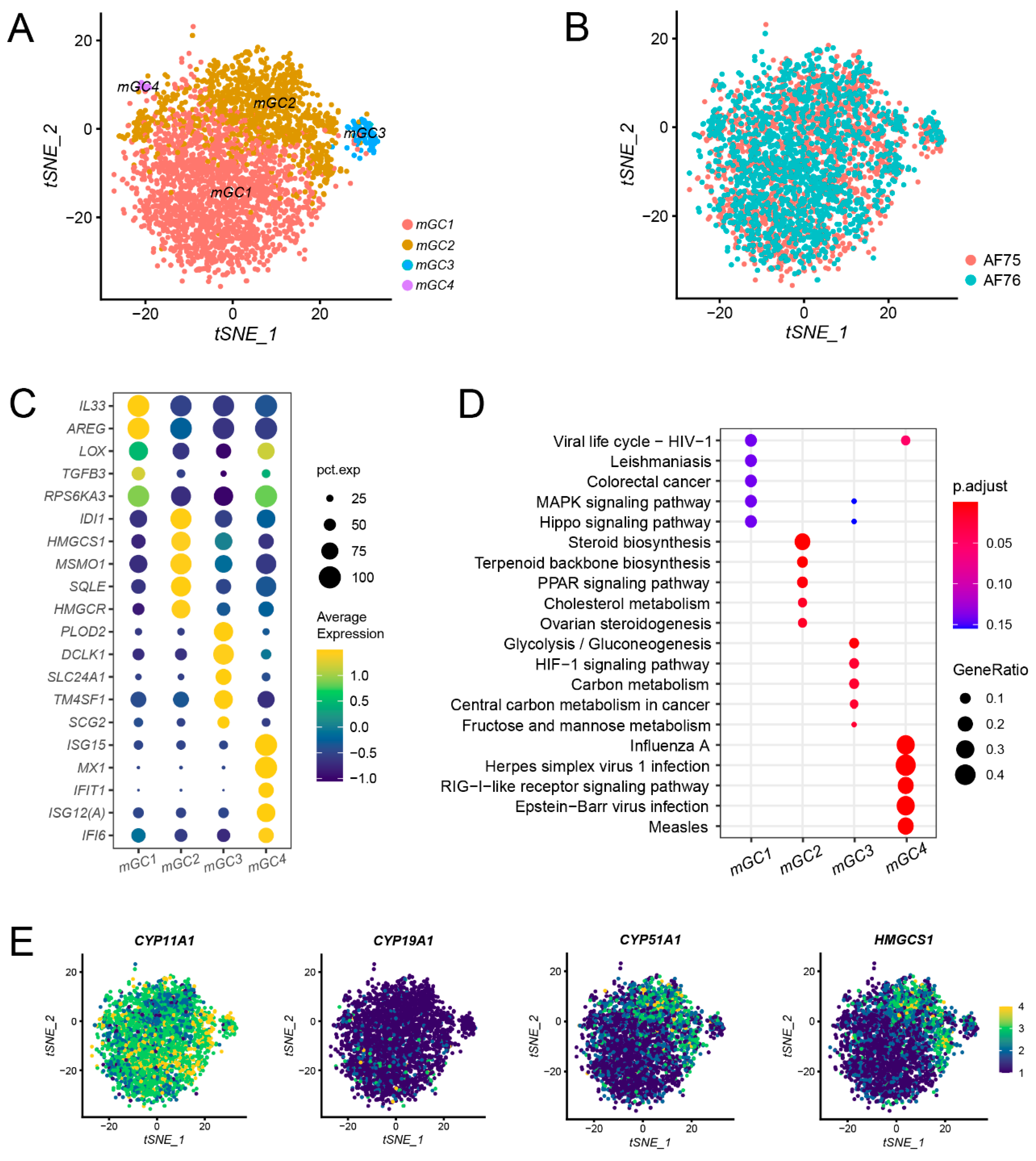
3.3. Subpopulation Identification of mGCs and the Expression of Steroid Synthesis-Associated Genes in mGCs
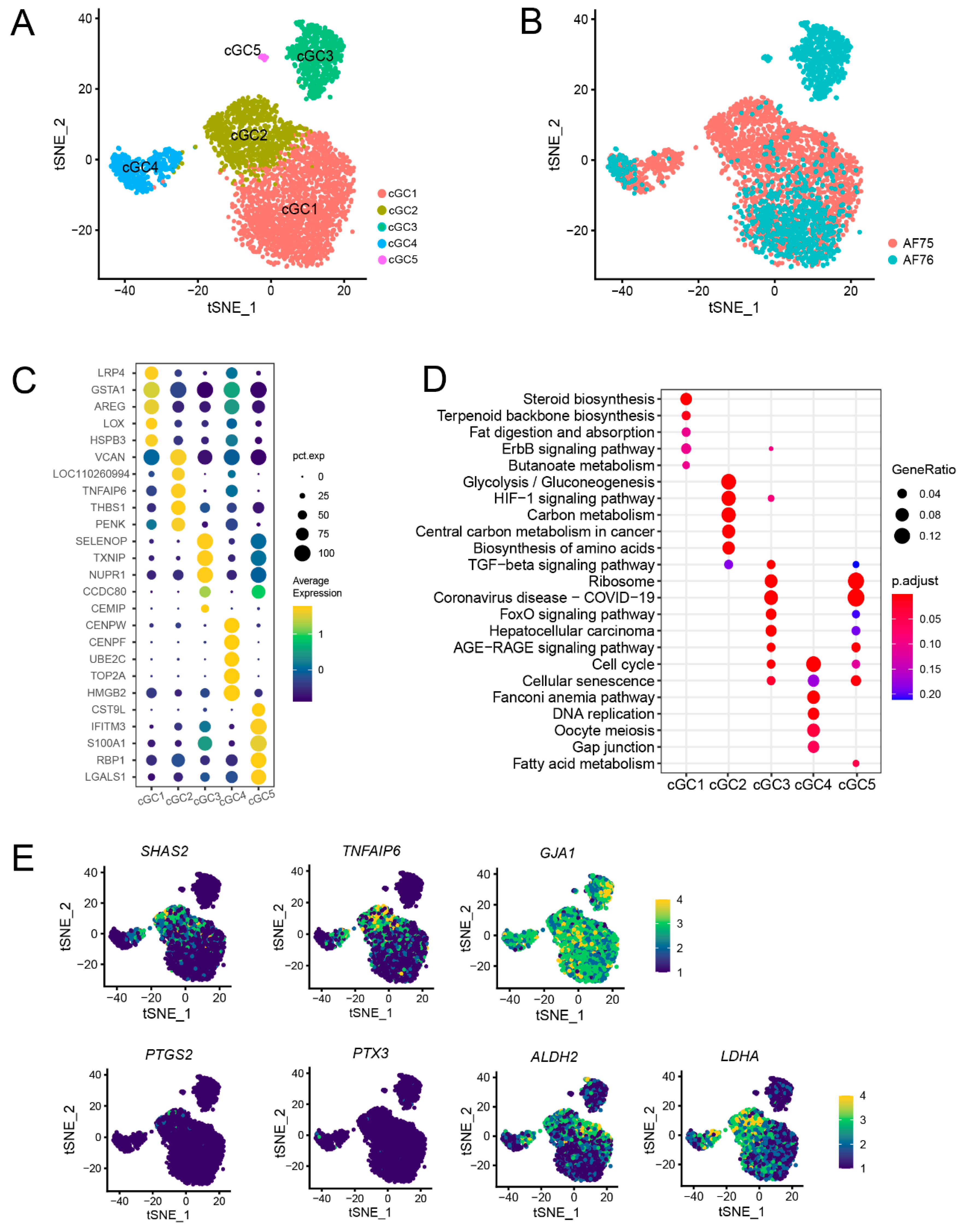
3.4. Subpopulations Identification and Annotation of cGCs Revealed a Difference in Metabolic Capacities between AF75 and AF76
3.5. Inter-Subpopulation Cell-Cell Communications among Granulosa Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van den Hurk, R.; Zhao, H. Formation of mammalian oocytes and their growth, differentiation and maturation within ovarian follicles. Theriogenology 2005, 63, 1717–1751. [Google Scholar] [CrossRef]
- Jaffe, L.A.; Egbert, J.R. Regulation of Mammalian Oocyte Meiosis by Intercellular Communication Within the Ovarian Follicle. Annu. Rev. Physiol. 2017, 79, 237–260. [Google Scholar] [CrossRef]
- Coticchio, G.; Dal Canto, M.; Mignini Renzini, M.; Guglielmo, M.C.; Brambillasca, F.; Turchi, D.; Novara, P.V.; Fadini, R. Oocyte maturation: Gamete-somatic cells interactions, meiotic resumption, cytoskeletal dynamics and cytoplasmic reorganization. Hum. Reprod. Update 2015, 21, 427–454. [Google Scholar] [CrossRef]
- Wigglesworth, K.; Lee, K.B.; Emori, C.; Sugiura, K.; Eppig, J.J. Transcriptomic diversification of developing cumulus and mural granulosa cells in mouse ovarian follicles. Biol. Reprod. 2015, 92, 23. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Bialecka, M.; Moustakas, I.; Lam, E.; Torrens-Juaneda, V.; Borggreven, N.V.; Trouw, L.; Louwe, L.A.; Pilgram, G.S.K.; Mei, H.; et al. Single-cell reconstruction of follicular remodeling in the human adult ovary. Nat. Commun. 2019, 10, 3164. [Google Scholar] [CrossRef]
- Li, S.; Chen, L.N.; Zhu, H.J.; Feng, X.; Xie, F.Y.; Luo, S.M.; Ou, X.H.; Ma, J.Y. Single-cell RNA sequencing analysis of mouse follicular somatic cellsdagger. Biol. Reprod. 2021, 105, 1234–1245. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Zhao, Y.; Ma, D.; Zhao, M.; Li, N.; Men, Y.; Zhang, Y.; Chu, H.; Lei, C.; et al. scRNA-seq of ovarian follicle granulosa cells from different fertility goats reveals distinct expression patterns. Reprod. Domest. Anim. 2021, 56, 801–811. [Google Scholar] [CrossRef]
- Lunney, J.K.; Van Goor, A.; Walker, K.E.; Hailstock, T.; Franklin, J.; Dai, C.H. Importance of the pig as a human biomedical model. Sci. Transl. Med. 2021, 13, eabd5758. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- McGinnis, C.S.; Murrow, L.M.; Gartner, Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019, 8, 329–337.e4. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Guerrero-Juarez, C.F.; Zhang, L.; Chang, I.; Ramos, R.; Kuan, C.H.; Myung, P.; Plikus, M.V.; Nie, Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Chen, N.; Feng, Y.; Li, N.; Pan, X.; Tian, Y.; Wang, J.; Jiang, Y.; He, D.; Li, J.; et al. Single-cell multi-omics profiling reveals key regulatory mechanisms that poise germinal vesicle oocytes for maturation in pigs. Cell. Mol. Life Sci. 2023, 80, 222. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Wang, S.; Zheng, Y.; Li, J.; Yu, Y.; Zhang, W.; Song, M.; Liu, Z.; Min, Z.; Hu, H.; Jing, Y.; et al. Single-Cell Transcriptomic Atlas of Primate Ovarian Aging. Cell 2020, 180, 585–600.e19. [Google Scholar] [CrossRef]
- Conti, M.; Hsieh, M.; Zamah, A.M.; Oh, J.S. Novel signaling mechanisms in the ovary during oocyte maturation and ovulation. Mol. Cell. Endocrinol. 2012, 356, 65–73. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.Y.; Sun, B.F.; Ma, Y.; Zhang, Y.; Wang, M.; Ma, C.; Shi, H.; Sun, Z.; Chen, J.; et al. Single-cell transcriptome profiling of the vaginal wall in women with severe anterior vaginal prolapse. Nat. Commun. 2021, 12, 87. [Google Scholar] [CrossRef]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef]
- Terenina, E.; Fabre, S.; Bonnet, A.; Monniaux, D.; Robert-Granié, C.; SanCristobal, M.; Sarry, J.; Vignoles, F.; Gondret, F.; Monget, P.; et al. Differentially expressed genes and gene networks involved in pig ovarian follicular atresia. Physiol. Genom. 2017, 49, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Moustakas, I.; Bialecka, M.; Del Valle, J.S.; Overeem, A.W.; Louwe, L.A.; Pilgram, G.S.K.; van der Westerlaken, L.A.J.; Mei, H.; Chuva de Sousa Lopes, S.M. Single-Cell Transcriptomics Analysis of Human Small Antral Follicles. Int. J. Mol. Sci. 2021, 22, 11955. [Google Scholar] [CrossRef]
- Dompe, C.; Kulus, M.; Stefanska, K.; Kranc, W.; Chermula, B.; Bryl, R.; Pienkowski, W.; Nawrocki, M.J.; Petitte, J.N.; Stelmach, B.; et al. Human Granulosa Cells-Stemness Properties, Molecular Cross-Talk and Follicular Angiogenesis. Cells 2021, 10, 1396. [Google Scholar] [CrossRef] [PubMed]
- Emori, C.; Kanke, T.; Ito, H.; Akimoto, Y.; Fujii, W.; Naito, K.; Sugiura, K. Expression and regulation of estrogen receptor 2 and its coregulators in mouse granulosa cells. J. Reprod. Dev. 2022, 68, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bonfiglio, R.; Banerji, S.; Jackson, D.; Salustri, A.; Richter, R. Micromechanical analysis of the hyaluronan-rich matrix surrounding the oocyte reveals a uniquely soft and elastic composition. Biophys. J. 2016, 110, 2779–2789. [Google Scholar] [CrossRef]
- Richani, D.; Dunning, K.R.; Thompson, J.G.; Gilchrist, R.B. Metabolic co-dependence of the oocyte and cumulus cells: Essential role in determining oocyte developmental competence. Hum. Reprod. Update 2021, 27, 27–47. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Li, Y.; Liu, X.; Wang, X.; Zhang, C.; Hao, C.; Deng, S. Regulation of follicular development and differentiation by intra-ovarian factors and endocrine hormones. Front. Biosci. (Landmark Ed.) 2019, 24, 983–993. [Google Scholar] [CrossRef]
- Boyer, A.; Goff, A.; Boerboom, D. WNT signaling in ovarian follicle biology and tumorigenesis. Trends Endocrinol. Metab. 2010, 21, 25–32. [Google Scholar] [CrossRef]
- Richani, D.; Gilchrist, R. The epidermal growth factor network: Role in oocyte growth, maturation and developmental competence. Hum. Reprod. Update 2018, 24, 1–14. [Google Scholar] [CrossRef]
- Wu, H.; Zhu, R.; Zheng, B.; Liao, G.; Wang, F.; Ding, J.; Li, H.; Li, M. Single-Cell Sequencing Reveals an Intrinsic Heterogeneity of the Preovulatory Follicular Microenvironment. Biomolecules 2022, 12, 231. [Google Scholar] [CrossRef]
- Miller, W.; Auchus, R. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Huang, J.; Zhang, D.; Xu, L.; Li, F.; Wu, L.; Liu, Z.; Zheng, Y. c-erbB2 and c-myb induce mouse oocyte maturation involving activation of maturation promoting factor. DNA Cell Biol. 2012, 31, 164–170. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, N.; Zhang, Y.; Tian, Y.; Wu, S.; Gao, F.; Yuan, X. Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell RNA Sequencing. Animals 2023, 13, 3019. https://doi.org/10.3390/ani13193019
Chen N, Zhang Y, Tian Y, Wu S, Gao F, Yuan X. Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell RNA Sequencing. Animals. 2023; 13(19):3019. https://doi.org/10.3390/ani13193019
Chicago/Turabian StyleChen, Na, Yong Zhang, Yuhan Tian, Shumei Wu, Fei Gao, and Xiaolong Yuan. 2023. "Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell RNA Sequencing" Animals 13, no. 19: 3019. https://doi.org/10.3390/ani13193019
APA StyleChen, N., Zhang, Y., Tian, Y., Wu, S., Gao, F., & Yuan, X. (2023). Deciphering Cellular Heterogeneity and Communication Patterns in Porcine Antral Follicles by Single-Cell RNA Sequencing. Animals, 13(19), 3019. https://doi.org/10.3390/ani13193019