
Vaginal Microbiome Dynamics of Cows in Different Parities

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal and Sample Collection
2.2. DNA Extraction
2.3. Polymerase Chain Reaction Amplification and Sequencing Library Construction
2.4. Taxonomic Classification and Statistical Analysis
3. Results
3.1. Number of Sequences
3.2. Classification of ASV
3.3. Community Structure of Vaginal Microorganisms and Changes in Correlation with Fecundity
3.4. Alpha Diversity Analysis
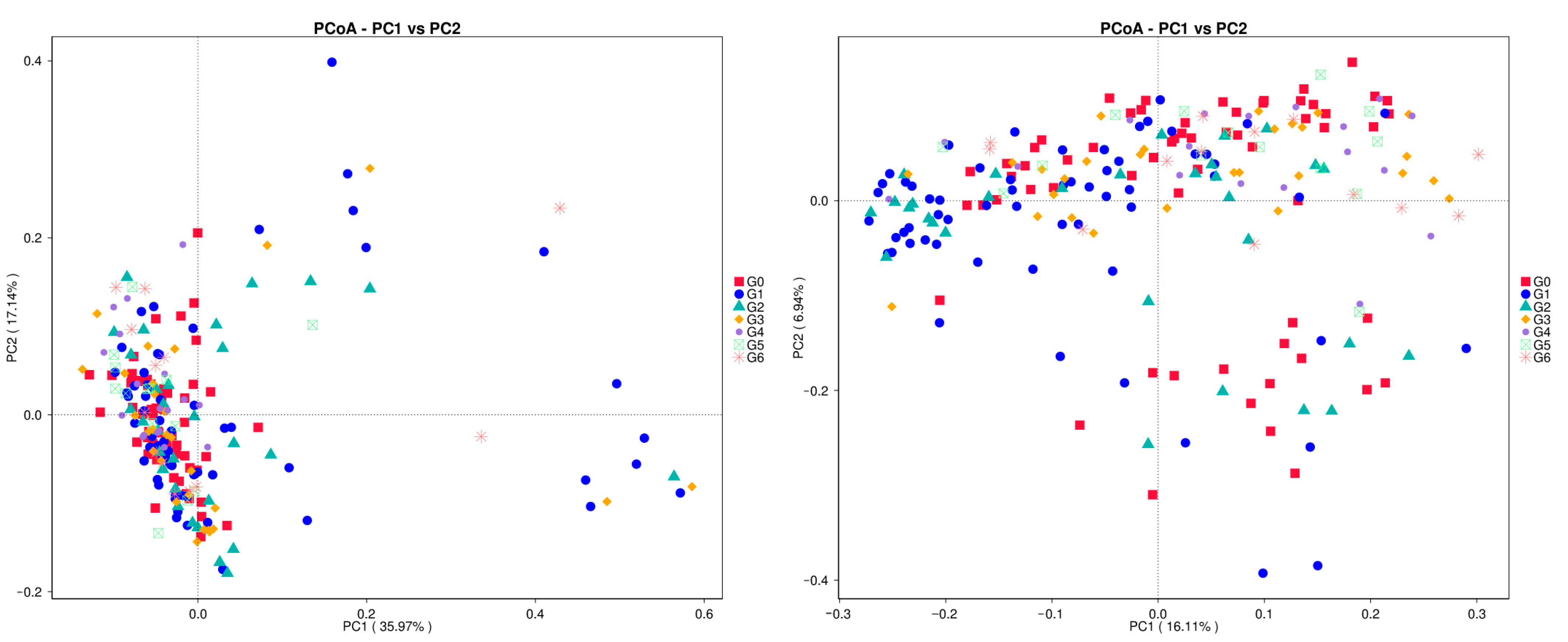
3.5. Beta Polymorphism Analysis
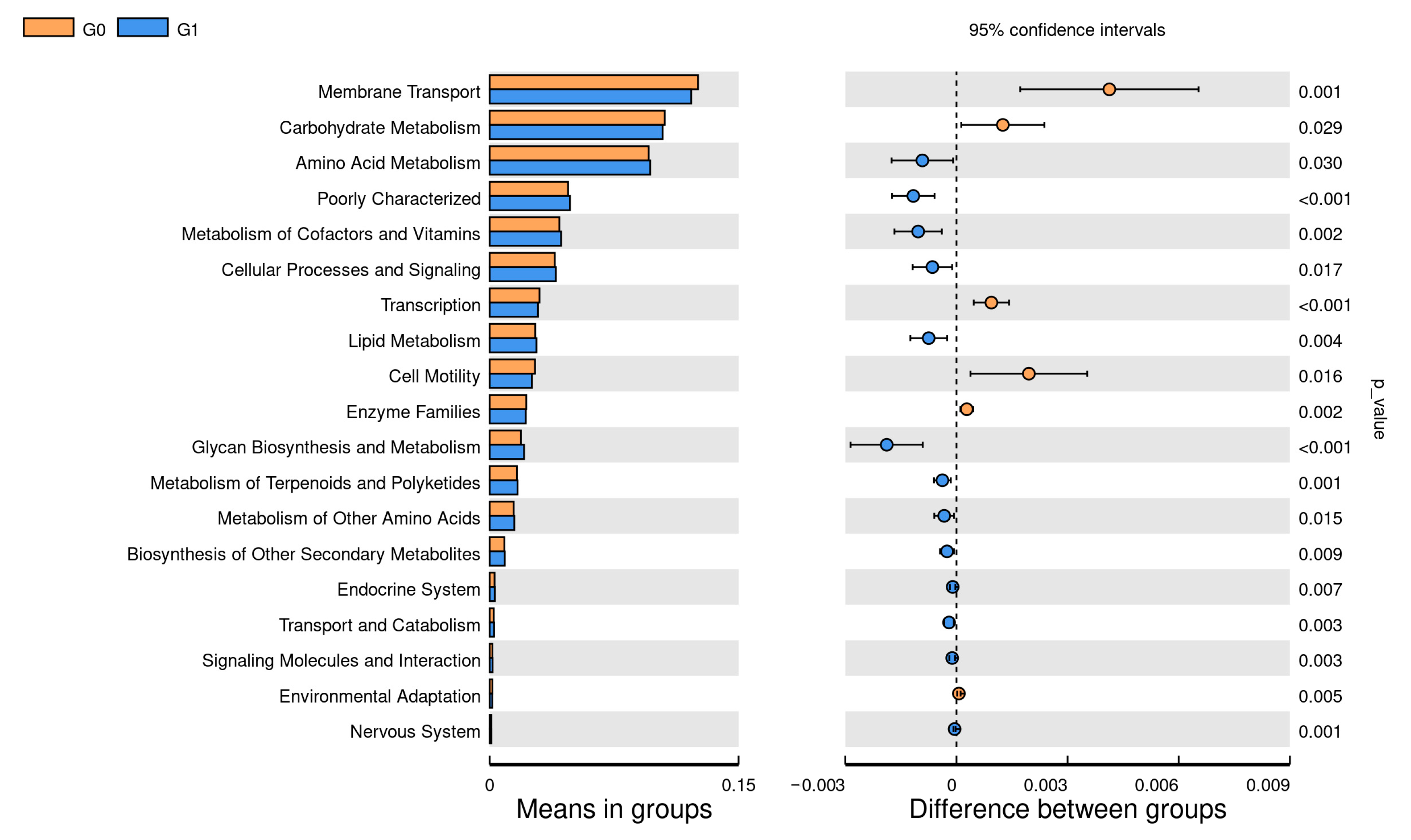
3.6. LEfSe
3.7. Association between Functional Properties of the Vaginal Microbiome and Age
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galvão, K.N.; Bicalho, R.C.; Jeon, S.J. Symposium review: The uterine microbiome associated with the development of uterine disease in dairy cows. J. Dairy Sci. 2019, 102, 11786–11797. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, I.M.; Cronin, J.G.; Bromfield, J.J. Tolerance and Innate Immunity Shape the Development of Postpartum Uterine Disease and the Impact of Endometritis in Dairy Cattle. Annu. Rev. Anim. Biosci. 2019, 7, 361–384. [Google Scholar] [CrossRef] [PubMed]
- Kovachev, S. Defence factors of vaginal lactobacilli. Crit. Rev. Microbiol. 2018, 44, 31–39. [Google Scholar] [CrossRef]
- Punzón-Jiménez, P.; Labarta, E. The impact of the female genital tract microbiome in women health and reproduction: A review. J. Assist. Reprod. Genet. 2021, 38, 2519–2541. [Google Scholar] [CrossRef]
- Machado, V.S.; Oikonomou, G.; Bicalho, M.L.; Knauer, W.A.; Gilbert, R.; Bicalho, R.C. Investigation of postpartum dairy cows’ uterine microbial diversity using metagenomic pyrosequencing of the 16S rRNA gene. Vet. Microbiol. 2012, 159, 460–469. [Google Scholar] [CrossRef]
- Ametaj, B.N.; Iqbal, S.; Selami, F.; Odhiambo, J.F.; Wang, Y.; Gänzle, M.G.; Dunn, S.M.; Zebeli, Q. Intravaginal administration of lactic acid bacteria modulated the incidence of purulent vaginal discharges, plasma haptoglobin concentrations, and milk production in dairy cows. Res. Vet. Sci. 2014, 96, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Odhiambo, J.F.; Farooq, U.; Lam, T.; Dunn, S.M.; Ametaj, B.N. Intravaginal lactic Acid bacteria modulated local and systemic immune responses and lowered the incidence of uterine infections in periparturient dairy cows. PLoS ONE 2014, 10, e0124167. [Google Scholar] [CrossRef]
- Owens, C.E.; Daniels, K.M.; Ealy, A.D.; Knowlton, K.F.; Cockrum, R.R. Graduate student literature review: Potential mechanisms of interaction between bacteria and the reproductive tract of dairy cattle. J. Dairy Sci. 2020, 103, 10951–10960. [Google Scholar] [CrossRef]
- Sheldon, I.M.; Price, S.B.; Cronin, J.; Gilbert, R.O.; Gadsby, J.E. Mechanisms of infertility associated with clinical and subclinical endometritis in high producing dairy cattle. Reprod. Domest. Anim. 2009, 44 (Suppl. S3), 1–9. [Google Scholar] [CrossRef]
- Sheldon, I.M.; Noakes, D.E.; Rycroft, A.N.; Pfeiffer, D.U.; Dobson, H. Influence of uterine bacterial contamination after parturition on ovarian dominant follicle selection and follicle growth and function in cattle. Reproduction 2002, 123, 837–845. [Google Scholar] [CrossRef]
- Pascottini, O.B.; Van Schyndel, S.J.; Spricigo, J.F.W.; Rousseau, J.; Weese, J.S.; LeBlanc, S.J. Dynamics of uterine microbiota in postpartum dairy cows with clinical or subclinical endometritis. Sci. Rep. 2020, 10, 12353. [Google Scholar] [CrossRef]
- Miranda-CasoLuengo, R.; Lu, J.; Williams, E.J.; Miranda-CasoLuengo, A.A.; Carrington, S.D.; Evans, A.C.O.; Meijer, W.G. Delayed differentiation of vaginal and uterine microbiomes in dairy cows developing postpartum endometritis. PLoS ONE 2019, 14, e0200974. [Google Scholar] [CrossRef] [PubMed]
- Ault, T.B.; Clemmons, B.A.; Reese, S.T.; Dantas, F.G.; Franco, G.A.; Smith, T.P.L.; Edwards, J.L.; Myer, P.R.; Pohler, K.G. Uterine and vaginal bacteria community diversity prior to artificial insemination between pregnant and nonpregnant postpartum cows1. J. Anim. Sci. 2019, 97, 4298–4304. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.L.; Liu, M.C.; Xu, J.; An, L.G.; Wang, J.F.; Zhu, Y.H. Uterine microbiota of dairy cows with clinical and subclinical endometritis. Front. Microbiol. 2018, 9, 2691. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, I.M.; Lewis, G.S.; LeBlanc, S.; Gilbert, R.O. Defining postpartum uterine disease in cattle. Theriogenology 2006, 65, 1516–1530. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.D. An update on pelvic inflammatory disease. Sex. Transm. Infect. 2002, 78, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Turni, C.; Blackall, P.J.; Boe-Hansen, G.; Hayes, B.J.; Tabor, A.E. Interrogating the bovine reproductive tract metagenomes using culture-independent approaches: A systematic review. Anim. Microbiome 2021, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Statist 1984, 11, 265–270. [Google Scholar]
- Pielou, E.C.; Pielou, E.C. Measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Li, B.; Zhang, X.; Guo, F.; Wu, W.; Zhang, T. Characterization of tetracycline resistant bacterial community in saline activated sludge using batch stress incubation with high-throughput sequencing analysis. Water Res. 2013, 47, 4207–4216. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, H.B. A generalization of Pielou’s index of non-randomness. J. Theor. Biol. 1979, 77, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.G.; Ericsson, A.C.; Poock, S.E.; Melendez, P.; Lucy, M.C. Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Pelzer, E.S.; Allan, J.A.; Waterhouse, M.A.; Ross, T.; Beagley, K.W.; Knox, C.L. Microorganisms within human follicular fluid: Effects on IVF. PLoS ONE 2013, 8, e59062. [Google Scholar] [CrossRef] [PubMed]
- Kudo, H.; Sugiura, T.; Higashi, S.; Oka, K.; Takahashi, M.; Kamiya, S.; Tamura, Y.; Usui, M. Characterization of reproductive microbiota of primiparous cows during early postpartum periods in the presence and absence of endometritis. Front. Vet. Sci. 2021, 8, 736996. [Google Scholar] [CrossRef]
- Rodrigues, N.F.; Kästle, J.; Coutinho, T.J.; Amorim, A.T.; Campos, G.B.; Santos, V.M.; Marques, L.M.; Timenetsky, J.; de Farias, S.T. Qualitative analysis of the vaginal microbiota of healthy cattle and cattle with genital-tract disease. Genet. Mol. Res. 2015, 14, 6518–6528. [Google Scholar] [CrossRef]
- Moore, S.G.; Ericsson, A.C.; Behura, S.K.; Lamberson, W.R.; Evans, T.J.; McCabe, M.S.; Poock, S.E.; Lucy, M.C. Concurrent and long-term associations between the endometrial microbiota and endometrial transcriptome in postpartum dairy cows. BMC Genom. 2019, 20, 405. [Google Scholar] [CrossRef]
- Williams, E.J.; Fischer, D.P.; Pfeiffer, D.U.; England, G.C.; Noakes, D.E.; Dobson, H.; Sheldon, I.M. Clinical evaluation of postpartum vaginal mucus reflects uterine bacterial infection and the immune response in cattle. Theriogenology 2005, 63, 102–117. [Google Scholar] [CrossRef]
- Bicalho, M.L.; Lima, F.S.; Machado, V.S.; Meira, E.B., Jr.; Ganda, E.K.; Foditsch, C.; Bicalho, R.C.; Gilbert, R.O. Associations among Trueperella pyogenes, endometritis diagnosis, and pregnancy outcomes in dairy cows. Theriogenology 2016, 85, 267–274. [Google Scholar] [CrossRef]
- Deng, F.; McClure, M.; Rorie, R.; Wang, X.; Chai, J.; Wei, X.; Lai, S.; Zhao, J. The vaginal and fecal microbiomes are related to pregnancy status in beef heifers. J. Anim. Sci. Biotechnol. 2019, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Amat, S.; Holman, D.B.; Schmidt, K.; Menezes, A.C.B.; Baumgaertner, F.; Winders, T.; Kirsch, J.D.; Liu, T.; Schwinghamer, T.D.; Sedivec, K.K.; et al. The Nasopharyngeal, Ruminal, and Vaginal Microbiota and the Core Taxa Shared across These Microbiomes in Virgin Yearling Heifers Exposed to Divergent In Utero Nutrition during Their First Trimester of Gestation and in Pregnant Beef Heifers in Response to Mineral Supplementation. Microorganisms 2021, 9, 2011. [Google Scholar] [CrossRef] [PubMed]
- Uchihashi, M.; Bergin, I.L.; Bassis, C.M.; Hashway, S.A.; Chai, D.; Bell, J.D. Influence of age, reproductive cycling status, and menstruation on the vaginal microbiome in baboons (Papio anubis). Am. J. Primatol. 2015, 77, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Q.; Li, L.X.; Zhao, H.Y.; Zhang, Y.H. Effects of parity and milk yield on lactation performance in dairy cows. South. J. Agric. Sci. 2022, 53, 2691–2700. [Google Scholar] [CrossRef]
- Moreno, I.; Codoñer, F.M.; Vilella, F.; Valbuena, D.; Martinez-Blanch, J.F.; Jimenez-Almazán, J.; Alonso, R.; Alamá, P.; Remohí, J.; Pellicer, A.; et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am. J. Obstet. Gynecol. 2016, 215, 684–703. [Google Scholar] [CrossRef] [PubMed]
- Messman, R.D.; Contreras-Correa, Z.E.; Paz, H.A.; Perry, G.; Lemley, C.O. Vaginal bacteria community composition and concentrations of estradiol at the time of artificial insemination in Brangus heifers. J. Anim. Sci. 2020, 98, skaa178. [Google Scholar] [CrossRef]
- Chee, W.J.Y.; Chew, S.Y.; Than, L.T.L. Vaginal microbiota and the potential of Lactobacillus derivatives in maintaining vaginal health. Microb. Cell Factories 2020, 19, 203. [Google Scholar] [CrossRef] [PubMed]
- Anahtar, M.N.; Byrne, E.H.; Doherty, K.E.; Bowman, B.A.; Yamamoto, H.S.; Soumillon, M.; Padavattan, N.; Ismail, N.; Moodley, A.; Sabatini, M.E.; et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 2015, 42, 965–976. [Google Scholar] [CrossRef]
- Chen, C.; Song, X.; Wei, W.; Zhong, H.; Dai, J.; Lan, Z.; Li, F.; Yu, X.; Feng, Q.; Wang, Z.; et al. The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 2017, 8, 875. [Google Scholar] [CrossRef]
- Giannattasio-Ferraz, S.; Laguardia-Nascimento, M.; Gasparini, M.R.; Leite, L.R.; Araujo, F.M.G.; de Matos Salim, A.C.; de Oliveira, A.P.; Nicoli, J.R.; de Oliveira, G.C.; da Fonseca, F.G.; et al. A common vaginal microbiota composition among breeds of Bos taurus indicus (Gyr and Nellore). Braz. J. Microbiol. 2019, 50, 1115–1124. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Shannon | Simpson | Chao1 | Pielou |
|---|---|---|---|---|
| G0 | 9.31 ± 0.35 ** | 0.995 ± 0.004 * | 1412.17 ± 174.69 ** | 0.89 ± 0.025 * |
| G1 | 8.40 ± 1.79 ** | 0.96 ± 0.08 * | 1226.41 ± 345.40 **ab | 0.82 ± 0.15 * |
| G2 | 8.75 ± 1.43 | 0.97 ± 0.06 | 1342.26 ± 302.81 | 0.84 ± 0.12 |
| G3 | 8.75 ± 1.69 | 0.97 ± 0.08 | 1382.55 ± 328.73 | 0.84 ± 0.14 |
| G4 | 9.24 ± 0.43 | 0.99 ± 0.004 | 1467.58 ± 219.76 b | 0.88 ± 0.03 |
| G5 | 9.29 ± 0.56 | 0.99 ± 0.01 | 1537.48 ± 161.72 a | 0.88 ± 0.05 |
| G6 | 8.79 ± 1.54 | 0.97 ± 0.065 | 1467.76 ± 269.76 | 0.84 ± 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, J.; Wang, J.; Zhao, K.; Chen, Y.; Xia, S.; Lai, S. Vaginal Microbiome Dynamics of Cows in Different Parities. Animals 2023, 13, 2880. https://doi.org/10.3390/ani13182880
Ni J, Wang J, Zhao K, Chen Y, Xia S, Lai S. Vaginal Microbiome Dynamics of Cows in Different Parities. Animals. 2023; 13(18):2880. https://doi.org/10.3390/ani13182880
Chicago/Turabian StyleNi, Jiale, Jie Wang, Kaisen Zhao, Yang Chen, Siqi Xia, and Songjia Lai. 2023. "Vaginal Microbiome Dynamics of Cows in Different Parities" Animals 13, no. 18: 2880. https://doi.org/10.3390/ani13182880
APA StyleNi, J., Wang, J., Zhao, K., Chen, Y., Xia, S., & Lai, S. (2023). Vaginal Microbiome Dynamics of Cows in Different Parities. Animals, 13(18), 2880. https://doi.org/10.3390/ani13182880