
Genetic Diversity of Porcine Circovirus Types 2 and 3 in Wild Boar in Italy

 ,
,  , , , ,
, , , ,  and
and 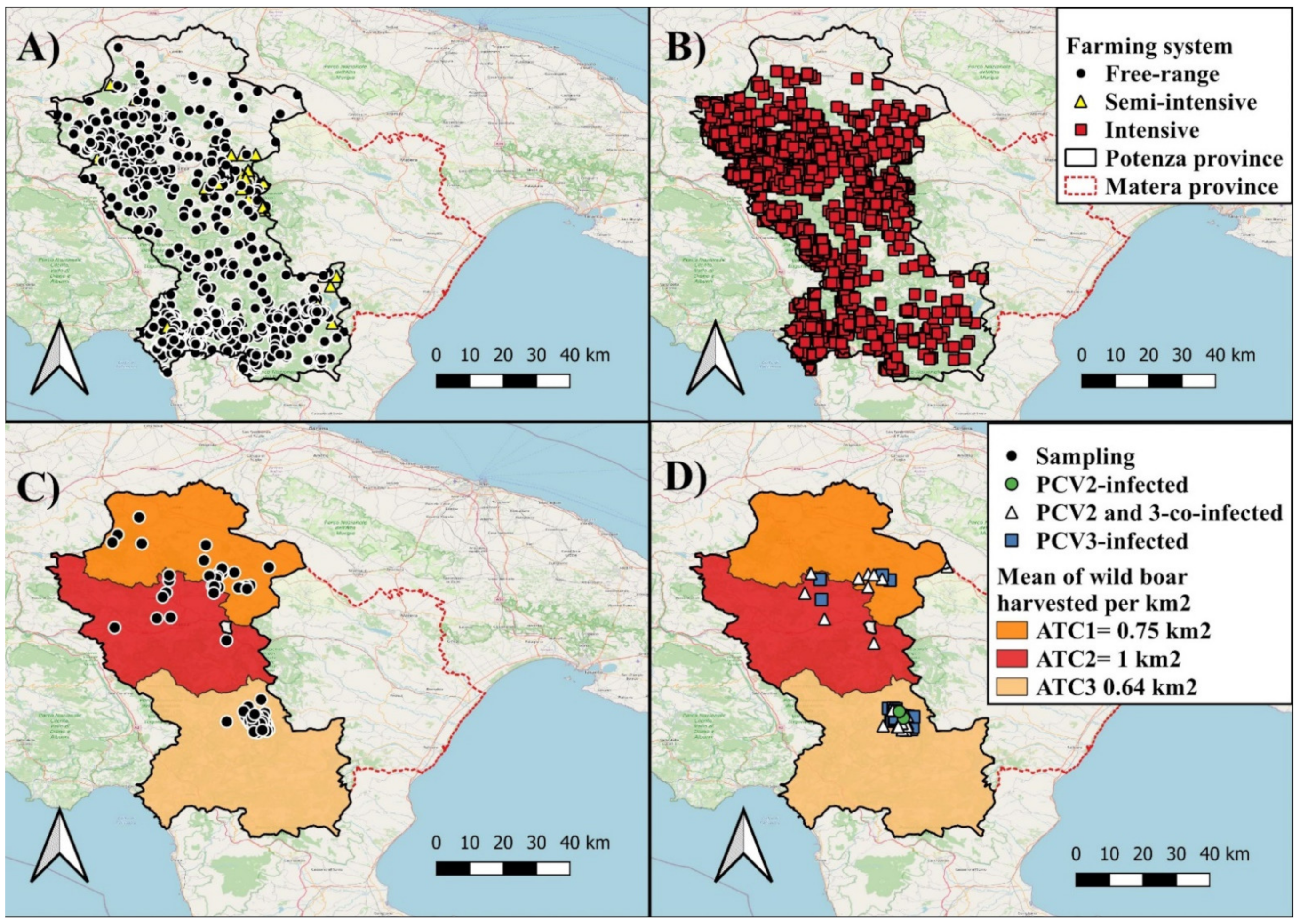{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Sampled Population
2.3. Nucleic Acids Extraction
2.4. Molecular Detection of PCV2 and PCV3
2.5. Strategy for Amplification of Complete Genomes of PCV2 and PCV3
2.6. Sequence and Phylogenetic Analyses
2.7. GenBank Sequence Submission
2.8. Statistical Analysis
3. Results
3.1. Epidemiology of PCVs
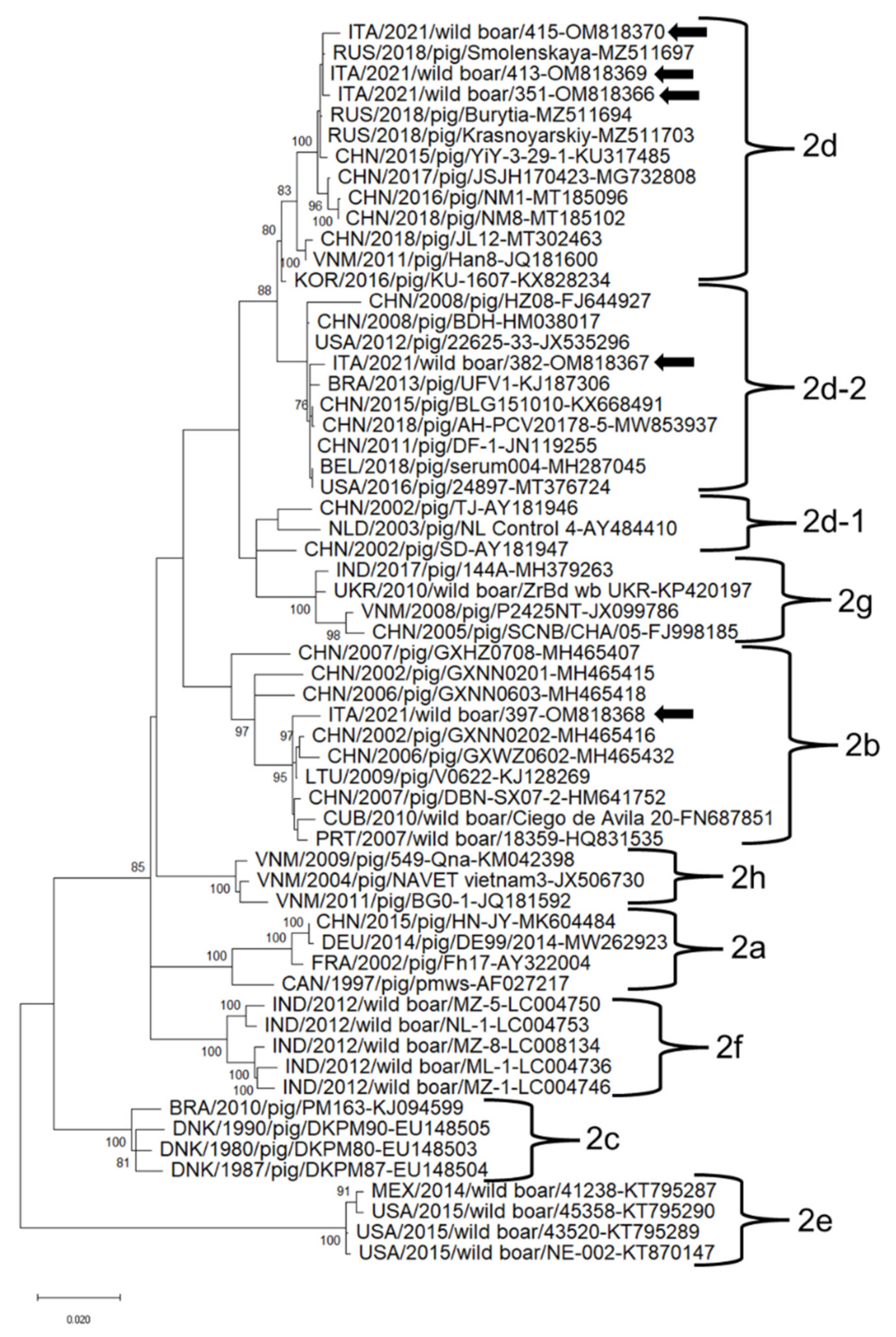
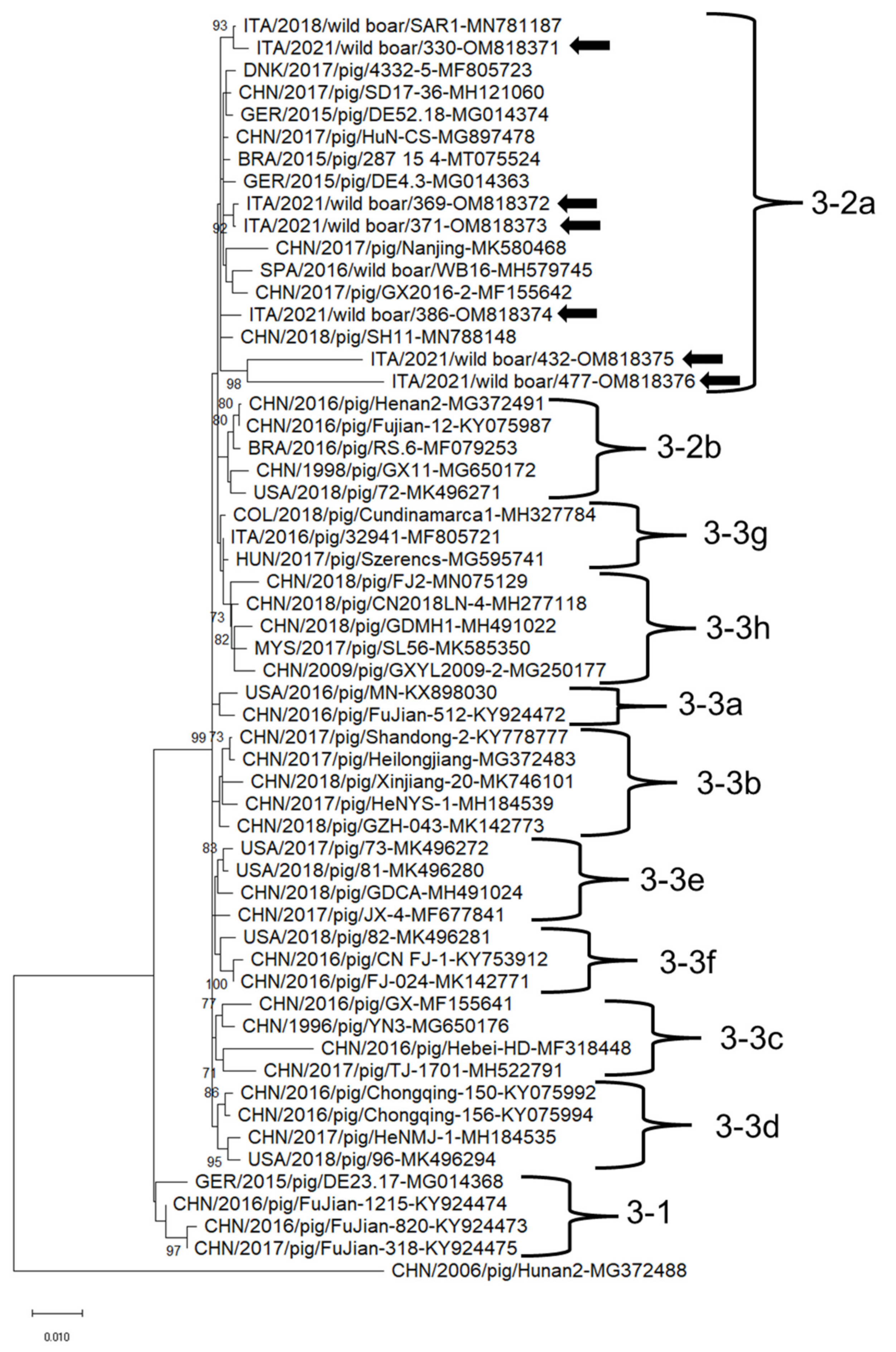
3.2. Sequence Analysis of PCVs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ellis, J. Porcine Circovirus: A Historical Perspective. Vet. Pathol. 2014, 51, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Cortey, M.; Olvera, A.; Novosel, D.; de Castro, A.M.M.G.; Biagini, P.; Segalés, J.; Drigo, M. Revisiting the Taxonomical Classification of Porcine Circovirus Type 2 (PCV2): Still a Real Challenge. Virol. J. 2015, 12, 131. [Google Scholar] [CrossRef] [PubMed]
- Tischer, I.; Gelderblom, H.; Vettermann, W.; Koch, M.A. A Very Small Porcine Virus with Circular Single-Stranded DNA. Nature 1982, 295, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Sirisereewan, C.; Thanawongnuwech, R.; Kedkovid, R. Current Understanding of the Pathogenesis of Porcine Circovirus 3. Pathogens 2022, 11, 1010064. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Hu, W.Q.; Li, J.Y.; Liu, T.N.; Zhou, J.Y.; Opriessnig, T.; Xiao, C.T. Novel Circovirus Species Identified in Farmed Pigs Designated as Porcine Circovirus 4, Hunan Province, China. Transbound. Emerg. Dis. 2020, 67, 1057–1061. [Google Scholar] [CrossRef]
- Franzo, G.; Ruiz, A.; Grassi, L.; Sibila, M.; Drigo, M.; Segalés, J. Lack of Porcine Circovirus 4 Genome Detection in Pig Samples from Italy and Spain. Pathogens 2020, 9, 433. [Google Scholar] [CrossRef]
- Allan, G.M.; McNeilly, F.; Cassidy, J.P.; Reilly, G.A.C.; Adair, B.; Ellis, W.A.; McNulty, M.S. Pathogenesis of Porcine Circovirus; Experimental Infections of Colostrum Deprived Piglets and Examination of Pig Foetal Material. Vet. Microbiol. 1995, 44, 49–64. [Google Scholar] [CrossRef]
- Klaumann, F.; Correa-Fiz, F.; Franzo, G.; Sibila, M.; Núñez, J.I.; Segalés, J. Current Knowledge on Porcine Circovirus 3 (PCV-3): A Novel Virus with a yet Unknown Impact on the Swine Industry. Front. Vet. Sci. 2018, 5, 315. [Google Scholar] [CrossRef]
- Palinski, R.; Piñeyro, P.; Shang, P.; Yuan, F.; Guo, R.; Fang, Y.; Byers, E.; Hause, B.M. A Novel Porcine Circovirus Distantly Related to Known Circoviruses is Associated with Porcine Dermatitis and Nephropathy Syndrome and Reproductive Failure. J. Virol. 2016, 91, e01879-16. [Google Scholar] [CrossRef]
- Ruiz-Fons, F.; Segalés, J.; Gortázar, C. A Review of Viral Diseases of the European Wild Boar: Effects of Population Dynamics and Reservoir Rôle. Vet. J. 2008, 176, 158–169. [Google Scholar] [CrossRef]
- Klaumann, F.; Dias-Alves, A.; Cabezón, O.; Mentaberre, G.; Castillo-Contreras, R.; López-Béjar, M.; Casas-Díaz, E.; Sibila, M.; Correa-Fiz, F.; Segalés, J. Porcine Circovirus 3 Is Highly Prevalent in Serum and Tissues and May Persistently Infect Wild Boar (Sus Scrofa Scrofa). Transbound. Emerg. Dis. 2019, 66, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Tinello, S.; Grassi, L.; Tucciarone, C.M.; Legnardi, M.; Cecchinato, M.; Dotto, G.; Mondin, A.; Martini, M.; Pasotto, D.; et al. Free to Circulate: An Update on the Epidemiological Dynamics of Porcine Circovirus 2 (PCV-2) in Italy Reveals the Role of Local Spreading, Wild Populations, and Foreign Countries. Pathogens 2020, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Lipej, Z.; Segalés, J.; Jemeršić, L.; Olvera, A.; Roić, B.; Novosel, D.; Mihaljević, Z.; Manojlović, L. First Description of Postweaning Multisystemic Wasting Syndrome (PMWS) in Wild Boar (Sus Scrofa) in Croatia and Phylogenetic Analysis of Partial PCV2 Sequences. Acta Vet. Hung. 2007, 55, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.; Billinis, C.; Psychas, V.; Birtsas, P.; Sofianidis, G.; Leontides, L.; Knowles, N.; Spyrou, V. Detection and Genetic Characterization of Porcine Circovirus 2 Isolates from the First Cases of Postweaning Multisystemic and Wasting Syndrome in Wild Boars in Greece. J. Wildl. Dis. 2008, 44, 864–870. [Google Scholar] [CrossRef]
- Amoroso, M.G.; Serra, F.; Esposito, C.; D’alessio, N.; Ferrara, G.; Cioffi, B.; Anzalone, A.; Pagnini, U.; de Carlo, E.; Fusco, G.; et al. Prevalence of Infection with Porcine Circovirus Types 2 and 3 in the Wild Boar Population in the Campania Region (Southern Italy). Animals 2021, 11, 3215. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Drigo, M.; Cecchinato, M.; Martini, M.; Mondin, A.; Menandro, M.L. First Report of Wild Boar Susceptibility to Porcine Circovirus Type 3: High Prevalence in the Colli Euganei Regional Park (Italy) in the Absence of Clinical Signs. Transbound. Emerg. Dis. 2018, 65, 957–962. [Google Scholar] [CrossRef]
- Dei Giudici, S.; Franzoni, G.; Bonelli, P.; Angioi, P.P.; Zinellu, S.; Deriu, V.; Carta, T.; Sechi, A.M.; Salis, F.; Balzano, F.; et al. Genetic Characterization of Porcine Circovirus 3 Strains Circulating in Sardinian Pigs and Wild Boars. Pathogens 2020, 9, 344. [Google Scholar] [CrossRef]
- Relun, A.; Grosbois, V.; Sánchez-Vizcaíno, J.M.; Alexandrov, T.; Feliziani, F.; Waret-Szkuta, A.; Molia, S.; Etter, E.M.C.; Martínez-López, B. Spatial and Functional Organization of Pig Trade in Different European Production Systems: Implications for Disease Prevention and Control. Front. Vet. Sci. 2016, 3, 4. [Google Scholar] [CrossRef]
- Cillis, G.; Statuto, D.; Picuno, P. Historical Gis as a Tool for Monitoring, Preserving and Planning Forest Landscape: A Case Study in a Mediterranean Region. Land 2021, 10, 851. [Google Scholar] [CrossRef]
- Valluzzi, C.; Rando, A.; Macciotta, N.P.P.; Gaspa, G.; di Gregorio, P. The Nero Lucano Pig Breed: Recovery and Variability. Animals 2021, 11, 1331. [Google Scholar] [CrossRef]
- Massei, G.; Toso, S. Biologia e Gestione Del Cinghialie; Spagnesi, M., Ed.; Ozzano, Istituto Nazionale Per La Fauna Selvatica: Bologna, Italy, 1993. [Google Scholar]
- Kim, H.R.; Park, Y.R.; Lim, D.R.; Park, M.J.; Park, J.Y.; Kim, S.H.; Lee, K.K.; Lyoo, Y.S.; Park, C.K. Multiplex Real-Time Polymerase Chain Reaction for the Differential Detection of Porcine Circovirus 2 and 3. J. Virol. Methods 2017, 250, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Franzo, G.; Segalés, J. Porcine Circovirus 2 (PCV-2) Genotype Update and Proposal of a New Genotyping Methodology. PLoS ONE 2018, 13, e0208585. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Nguyen, V.G.; Park, Y.H.; Park, B.K. Genotyping of PCV3 Based on Reassembled Viral Gene Sequences. Vet. Med. Sci. 2021, 7, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Müller, H.; Rector, A.; van Ranst, M.; Stevens, H. Rolling-Circle Amplification of Viral DNA Genomes Using Phi29 Polymerase. Trends Microbiol. 2009, 17, 205–211. [Google Scholar] [CrossRef]
- Rector, A.; Tachezy, R.; van Ranst, M. A Sequence-Independent Strategy for Detection and Cloning of Circular DNA Virus Genomes by Using Multiply Primed Rolling-Circle Amplification. J. Virol. 2004, 78, 4993–4998. [Google Scholar] [CrossRef]
- Beikpour, F.; Ndiana, L.A.; Sazmand, A.; Capozza, P.; Nemati, F.; Pellegrini, F.; Zafari, S.; Zolhavarieh, S.M.; Cardone, R.; Faraji, R.; et al. Detection and Genomic Characterization of Canine Circovirus in Iran. Animals 2022, 12, 507. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3066. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Agresti, A.; Coull, B.A. Approximate Is Better than “Exact” for Interval Estimation of Binomial Proportions. Am. Stat. 1998, 52, 119–126. [Google Scholar]
- Nieves, E.; Jones, J. Epi Info™: Now an Open-source application that continues a long and productive “life” through CDC support and funding. Pan. Afr. Med. J. 2009, 2, 6. [Google Scholar]
- Raev, S.; Yuzhakov, A.; Aliper, T.; Franzo, G. Whole-Genome Analysis of Porcine Circovirus Type 2 in Russia. Pathogens 2021, 10, 1631. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Xie, J.; Theuns, S.; Nauwynck, H.J. Changes on the Viral Capsid Surface during the Evolution of Porcine Circovirus Type 2 (PCV2) from 2009 till 2018 May Lead to a Better Receptor Binding. Virus Evol. 2019, 5, vez026. [Google Scholar] [CrossRef] [PubMed]
- Nainys, J.; Lasickiene, R.; Petraityte-burneikiene, R.; Dabrisius, J.; Lelesius, R.; Sereika, V.; Zvirbliene, A.; Sasnauskas, K.; Gedvilaite, A. Generation in Yeast of Recombinant Virus-like Particles of Porcine Circovirus Type 2 Capsid Protein and Their Use for a Serologic Assay and Development of Monoclonal Antibodies. BMC Biotechnol. 2014, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liu, X.; Zhang, P.; Wang, L.; Liu, Y.; Zhang, L.; Liang, P.; Song, C. Genome Characterization of a Porcine Circovirus Type 3 in South China. Transbound. Emerg. Dis. 2018, 65, 264–266. [Google Scholar] [CrossRef]
- Wang, T.; Chai, W.; Wang, Y.; Liu, W.; Huang, Z.; Chen, L.; Guo, R.; Dong, Y.; Liu, M.; Zheng, Q.; et al. First Detection and Phylogenetic Analysis of Porcine Circovirus 3 in Female Donkeys with Reproductive Disorders. BMC Vet. Res. 2021, 17, 308. [Google Scholar] [CrossRef]
- Saporiti, V.; Huerta, E.; Correa-Fiz, F.; Grosse Liesner, B.; Duran, O.; Segalés, J.; Sibila, M. Detection and Genotyping of Porcine Circovirus 2 (PCV-2) and Detection of Porcine Circovirus 3 (PCV-3) in Sera from Fattening Pigs of Different European Countries. Transbound. Emerg. Dis. 2020, 67, 2531. [Google Scholar] [CrossRef]
- Cságola, A.; Kecskeméti, S.; Kardos, G.; Kiss, I.; Tuboly, T. Genetic Characterization of Type 2 Porcine Circoviruses Detected in Hungarian Wild Boars. Arch. Virol. 2005, 151, 495–507. [Google Scholar] [CrossRef]
- Hammer, R.; Ritzmann, M.; Palzer, A.; Lang, C.; Hammer, B.; Pesch, S.; Ladinig, A. Porcine Reproductive and Respiratory Syndrome Virus and Porcine Circovirus Type 2 Infections in Wild Boar (Sus Scrofa) in Southwestern Germany. J. Wildl. Dis. 2012, 48, 87–094. [Google Scholar] [CrossRef]
- Sliz, I.; Vlasakova, M.; Jackova, A.; Vilcek, S. Characterization of Porcine Parvovirus Type 3 and Porcine Circovirus Type 2 in Wild Boars (Sus Scrofa) in Slovakia. J. Wildl. Dis. 2015, 51, 703–711. [Google Scholar] [CrossRef]
- Bhide, K.; Csank, T.; Pistl, J.; Ciberej, J. Prevalence of Porcine Circovirus 2 and Virus-Specific Antibodies in Wild Boars (Sus Scrofa) in Slovakia. Acta Virol. 2014, 58, 386–388. [Google Scholar] [CrossRef]
- Nisavic, J.; Milic, N.; Radalj, A.; Mirilovic, M.; Vejnovic, B.; Cosic, M.; Knezevic, A.; Veljovic, L.; Zivulj, A. Detection and Characterisation of Porcine Circoviruses in Wild Boars in Northeastern Serbia. Veterinární Med. 2022, 67, 131–137. [Google Scholar] [CrossRef]
- Henriques, A.M.; Duarte, M.; Fagulha, T.; Ramos, F.; Barros, S.C.; Luís, T.; Fevereiro, M. Molecular Study of Porcine Circovirus Type 2 Circulating in Portugal. Infect. Genet. Evol. 2011, 11, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, M.; Szczotka, A.; Podgórska, K.; Stadejek, T. Prevalence of Infection and Genetic Diversity of Porcine Circovirus Type 2 (PCV2) in Wild Boar (Sus Scrofa) in Poland. J. Wildl. Dis. 2012, 48, 612–618. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cadar, D.; Cságola, A.; Spinu, M.; Dán, D.; Ursu, K.; Lorincz, M.; Tuboly, T. Prevalence of Porcine Circoviruses in Transylvanian Wild Boars, Detected by Real-Time PCR—Short Communication. Acta Vet. Hung. 2010, 58, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Dei Giudici, S.; D’Avino, C.; Salaris, A.A.; Sulas, A.; Madrau, M.P.; Sanna, M.L.; Oggiano, A. Caratterizzazione Molecolare Di PCV2 Nei Suini Selvatici e Domestici in Sardegna. In Proceedings of the XIV Congresso Nazionale S.I.Di.L.V., Sorrento, Italy, 24–26 October 2012. [Google Scholar]
- Mur, L.; Sánchez-Vizcaíno, J.M.; Fernández-Carrión, E.; Jurado, C.; Rolesu, S.; Feliziani, F.; Laddomada, A.; Martínez-López, B. Understanding African Swine Fever Infection Dynamics in Sardinia Using a Spatially Explicit Transmission Model in Domestic Pig Farms. Transbound. Emerg. Dis. 2018, 65, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, M.; Halbur, P.G.; Haqshenas, G.; Royer, R.; Thomas, P.; Nawagitgul, P.; Gill, M.; Toth, T.E.; Meng, X.J. Cloned Genomic DNA of Type 2 Porcine Circovirus is Infectious When Injected Directly into the Liver and Lymph Nodes of Pigs: Characterization of Clinical Disease, Virus Distribution, and Pathologic Lesions. J. Virol. 2002, 76, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Yoo, S.J.; Park, C.K.; Lyoo, Y.S. Prevalence of Novel Porcine Circovirus 3 in Korean Pig Populations. Vet. Microbiol. 2017, 207, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Klaumann, F.; Franzo, G.; Sohrmann, M.; Correa-Fiz, F.; Drigo, M.; Núñez, J.I.; Sibila, M.; Segalés, J. Retrospective Detection of Porcine Circovirus 3 (PCV-3) in Pig Serum Samples from Spain. Transbound. Emerg. Dis. 2018, 65, 1290–1296. [Google Scholar] [CrossRef]
- Grau-Roma, L.; Hjulsager, C.K.; Sibila, M.; Kristensen, C.S.; López-Soria, S.; Enøe, C.; Casal, J.; Bøtner, A.; Nofrarías, M.; Bille-Hansen, V.; et al. Infection, Excretion and Seroconversion Dynamics of Porcine Circovirus Type 2 (PCV2) in Pigs from Post-Weaning Multisystemic Wasting Syndrome (PMWS) Affected Farms in Spain and Denmark. Vet. Microbiol. 2009, 135, 272–282. [Google Scholar] [CrossRef]
- Eddicks, M.; Beuter, B.; Stuhldreier, R.; Nolte, T.; Reese, S.; Sutter, G.; Ritzmann, M.; Fux, R. Cross-Sectional Study on Viraemia and Shedding of Porcine Circovirus Type 2 in a Subclinically Infected Multiplier Sow Herd. Vet. Rec. 2019, 184, 189. [Google Scholar] [CrossRef]
- Eddicks, M.; Müller, M.; Fux, R.; Ritzmann, M.; Stadler, J. Detection of Porcine Circovirus Type 3 DNA in Serum and Semen Samples of Boars from a German Boar Stud. Vet. J. 2022, 279, 105784. [Google Scholar] [CrossRef] [PubMed]
- Dei Giudici, S.; lo Presti, A.; Bonelli, P.; Angioi, P.P.; Sanna, G.; Zinellu, S.; Balzano, F.; Salis, F.; Ciccozzi, M.; Oggiano, A. Phylogenetic Analysis of Porcine Circovirus Type 2 in Sardinia, Italy, Shows Genotype 2d Circulation among Domestic Pigs and Wild Boars. Infect. Genet. Evol. 2019, 71, 189–196. [Google Scholar] [CrossRef]
- Truvé, J.; Lemel, J. Timing and Distance of Natal Dispersal for Wild Boar Sus Scrofa in Sweden. Wildl. Biol. 2003, 9, 51–57. [Google Scholar] [CrossRef]
- Veličković, N.; Ferreira, E.; Djan, M.; Ernst, M.; Obreht Vidaković, D.; Monaco, A.; Fonseca, C. Demographic History, Current Expansion and Future Management Challenges of Wild Boar Populations in the Balkans and Europe. Heredity 2016, 117, 348. [Google Scholar] [CrossRef] [PubMed]
- Maistrelli, C.; Hüneke, H.; Langeheine, M.; Keuling, O.; Siebert, U.; Brehm, R. Precocious Puberty in Male Wild Boars: A Possible Explanation for the Dramatic Population Increase in Germany and Europe. PeerJ 2021, 9, e11798. [Google Scholar] [CrossRef] [PubMed]
- Mysterud, A.; Rivrud, I.M.; Gundersen, V.; Rolandsen, C.M.; Viljugrein, H. The Unique Spatial Ecology of Human Hunters. Nat. Hum. Behav. 2020, 4, 694–701. [Google Scholar] [CrossRef]
- Fanelli, A.; Perrone, A.; Ferroglio, E. Spatial and Temporal Dynamics of Wild Boars Sus Scrofa Hunted in Alpine Environment. Eur. J. Wildl. Res. 2021, 67, 47. [Google Scholar] [CrossRef]
- Ryser-Degiorgis, M.P. Wildlife Health Investigations: Needs, Challenges and Recommendations. BMC Vet. Res. 2013, 9, 223. [Google Scholar] [CrossRef]
- Fanelli, A.; Tizzani, P.; Ferroglio, E.; Belleau, E. Cheilospirura Hamulosa in the Rock Partridge (Alectoris Graeca Saxatilis): Epidemiological Patterns and Prediction of Parasite Distribution in France. Diversity 2020, 12, 484. [Google Scholar] [CrossRef]
- Gontero, C.; Fanelli, A.; Zanet, S.; Meneguz, P.G.; Tizzani, P. Exotic Species and Autochthonous Parasites: Trichostrongylus Retortaeformis in Eastern Cottontail. Life 2020, 10, 31. [Google Scholar] [CrossRef]
- Sauter-Louis, C.; Conraths, F.J.; Probst, C.; Blohm, U.; Schulz, K.; Sehl, J.; Fischer, M.; Forth, J.H.; Zani, L.; Depner, K.; et al. African Swine Fever in Wild Boar in Europe—A Review. Viruses 2021, 13, 1717. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanelli, A.; Pellegrini, F.; Camero, M.; Catella, C.; Buonavoglia, D.; Fusco, G.; Martella, V.; Lanave, G. Genetic Diversity of Porcine Circovirus Types 2 and 3 in Wild Boar in Italy. Animals 2022, 12, 953. https://doi.org/10.3390/ani12080953
Fanelli A, Pellegrini F, Camero M, Catella C, Buonavoglia D, Fusco G, Martella V, Lanave G. Genetic Diversity of Porcine Circovirus Types 2 and 3 in Wild Boar in Italy. Animals. 2022; 12(8):953. https://doi.org/10.3390/ani12080953
Chicago/Turabian StyleFanelli, Angela, Francesco Pellegrini, Michele Camero, Cristiana Catella, Domenico Buonavoglia, Giovanna Fusco, Vito Martella, and Gianvito Lanave. 2022. "Genetic Diversity of Porcine Circovirus Types 2 and 3 in Wild Boar in Italy" Animals 12, no. 8: 953. https://doi.org/10.3390/ani12080953
APA StyleFanelli, A., Pellegrini, F., Camero, M., Catella, C., Buonavoglia, D., Fusco, G., Martella, V., & Lanave, G. (2022). Genetic Diversity of Porcine Circovirus Types 2 and 3 in Wild Boar in Italy. Animals, 12(8), 953. https://doi.org/10.3390/ani12080953