
Analysis Polyadenylation Signal Usage in Sus scrofa

Abstract
:Simple Summary
Abstract
1. Introduction
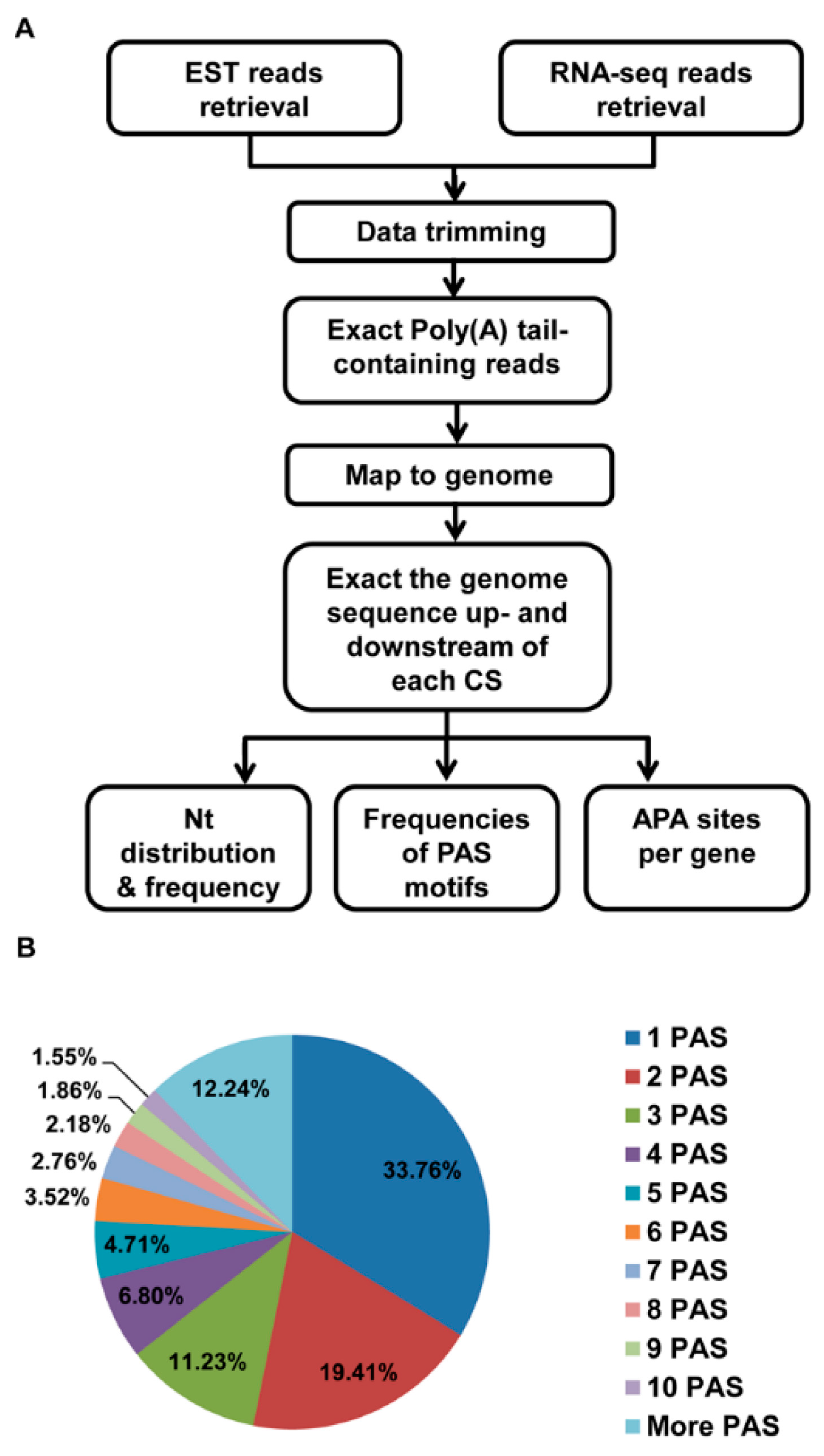
2. Materials and Methods
2.1. The Source of the EST Data and RNA Sequence Data
2.2. RNA Sequence Data Trimming
2.3. Identification of Poly(A) Sites in the EST and RNA-Seq Data
2.4. Polyadenylation Signal Search and Analysis
2.5. APA Analysis
2.6. PAS Annotation
2.7. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
3. Results
3.1. Identification of pA Sites
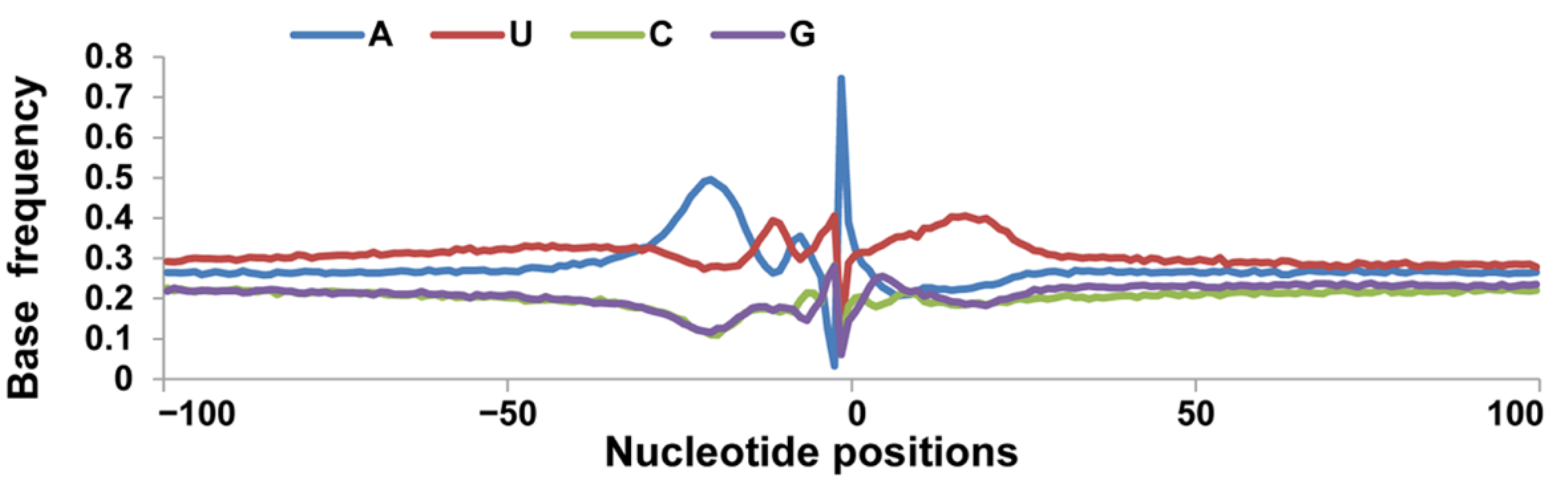
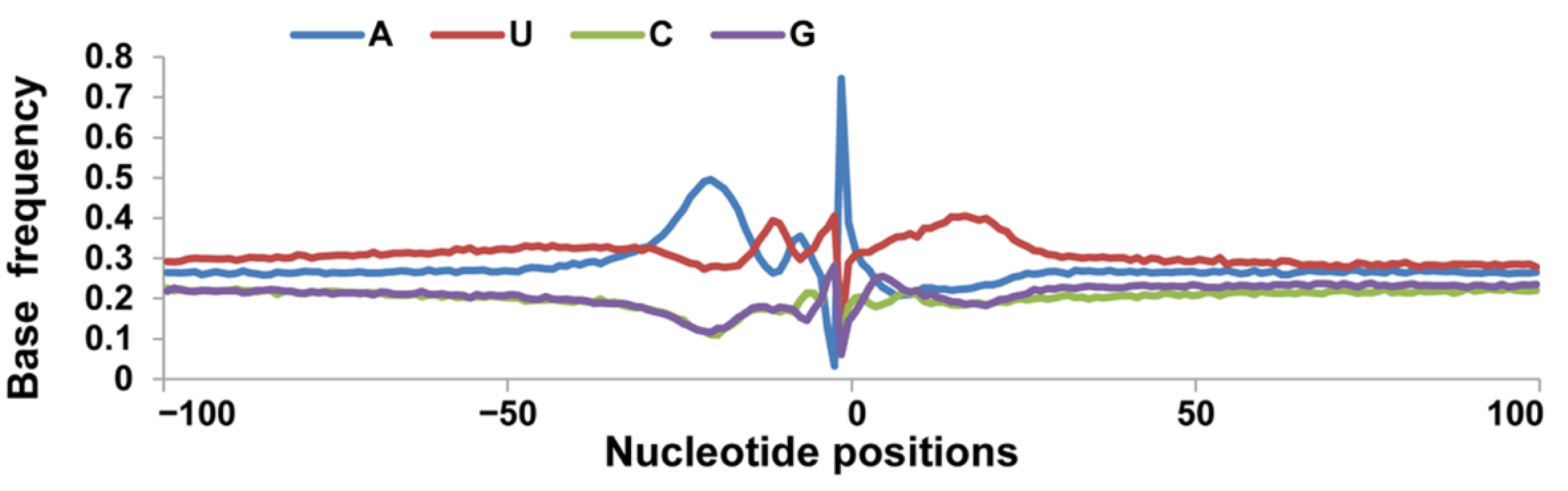
3.2. Nucleotide Bias near the Cleavage Sites
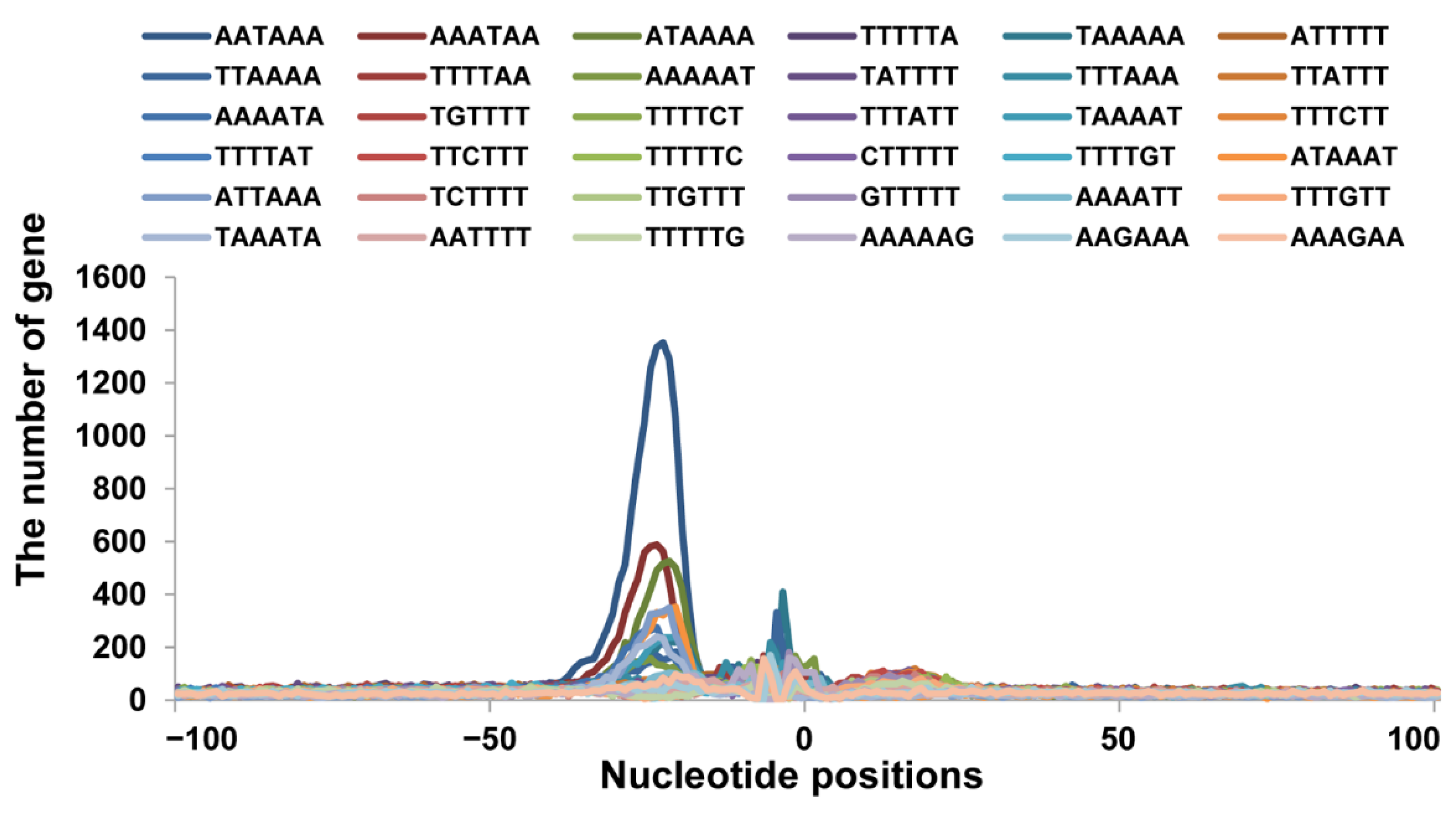
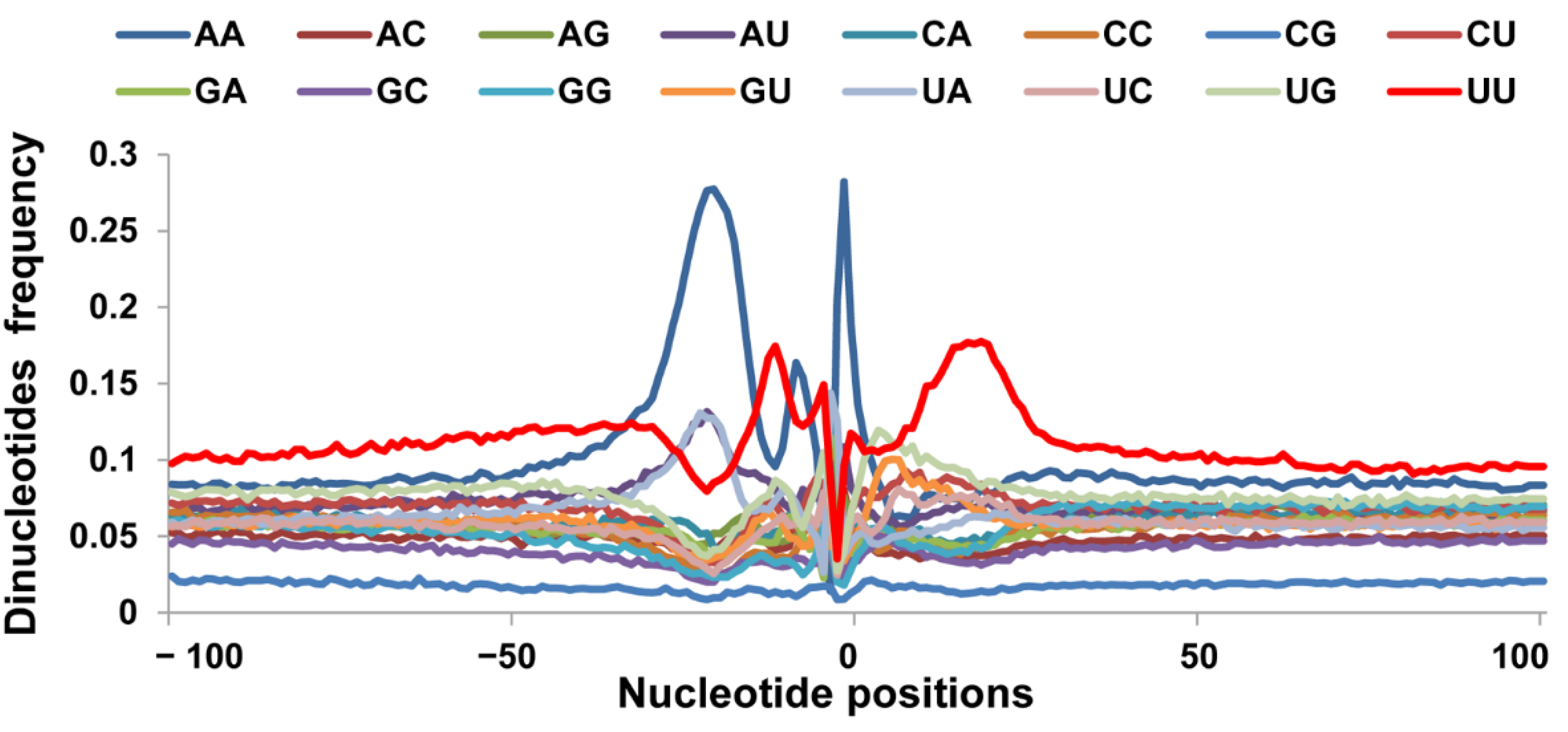
3.3. Sequence Motifs near the Cleavage Sites
3.4. Usage Frequencies of AAUAAA and Single Nucleotide Variants Polyadenylation Signal Variants
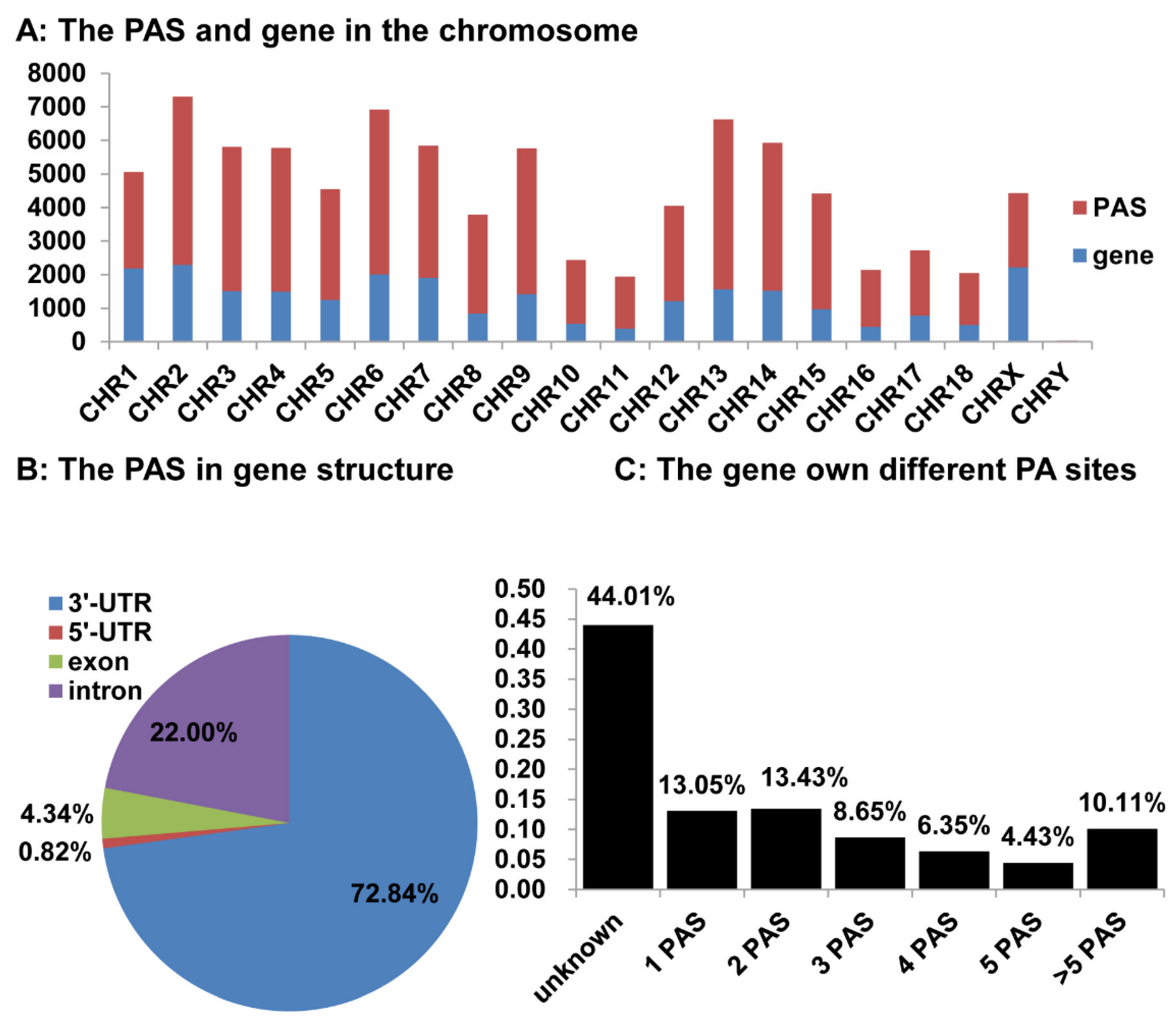
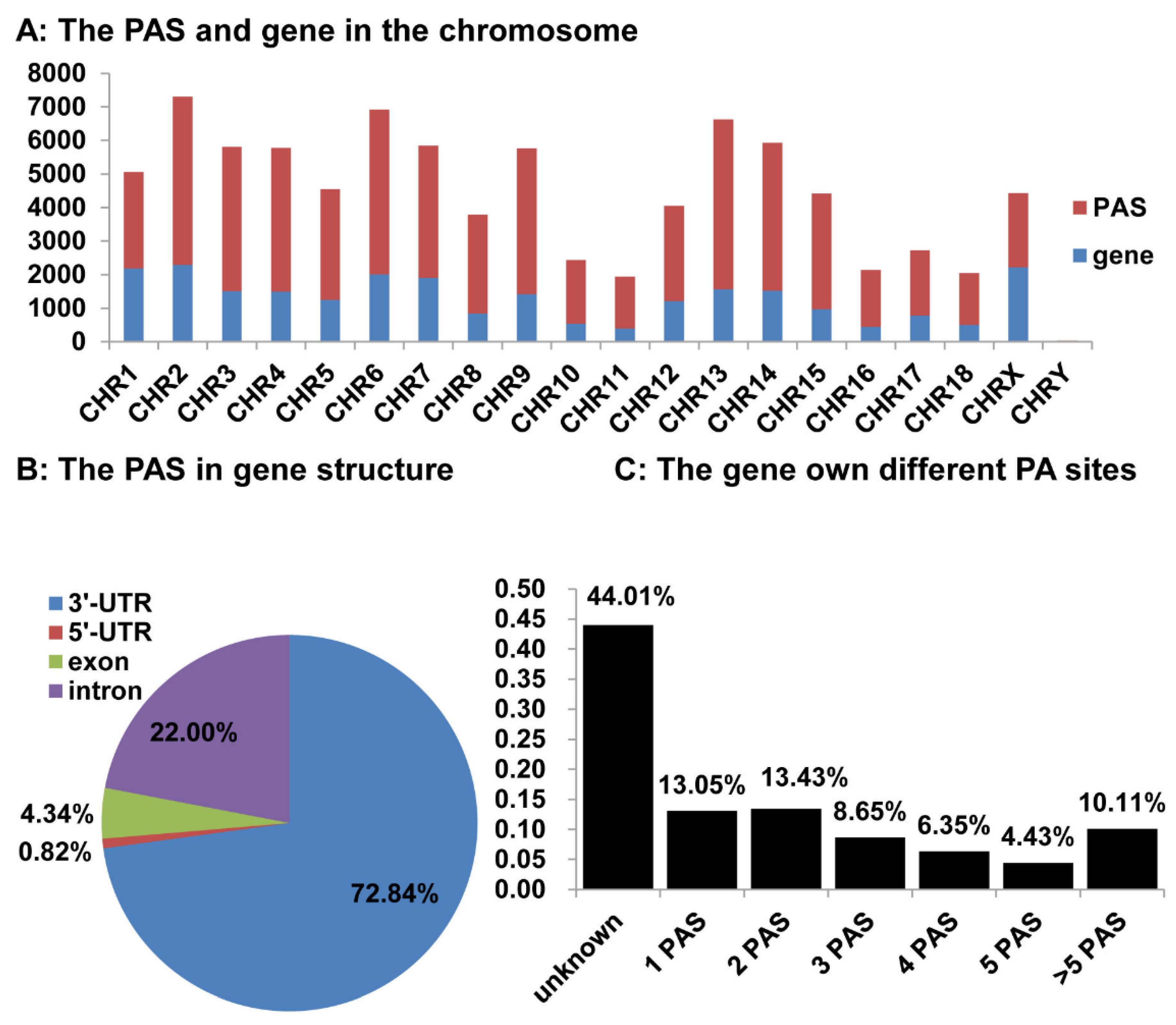
3.5. Distribution of the Identified pA Sites in the Pig Genome
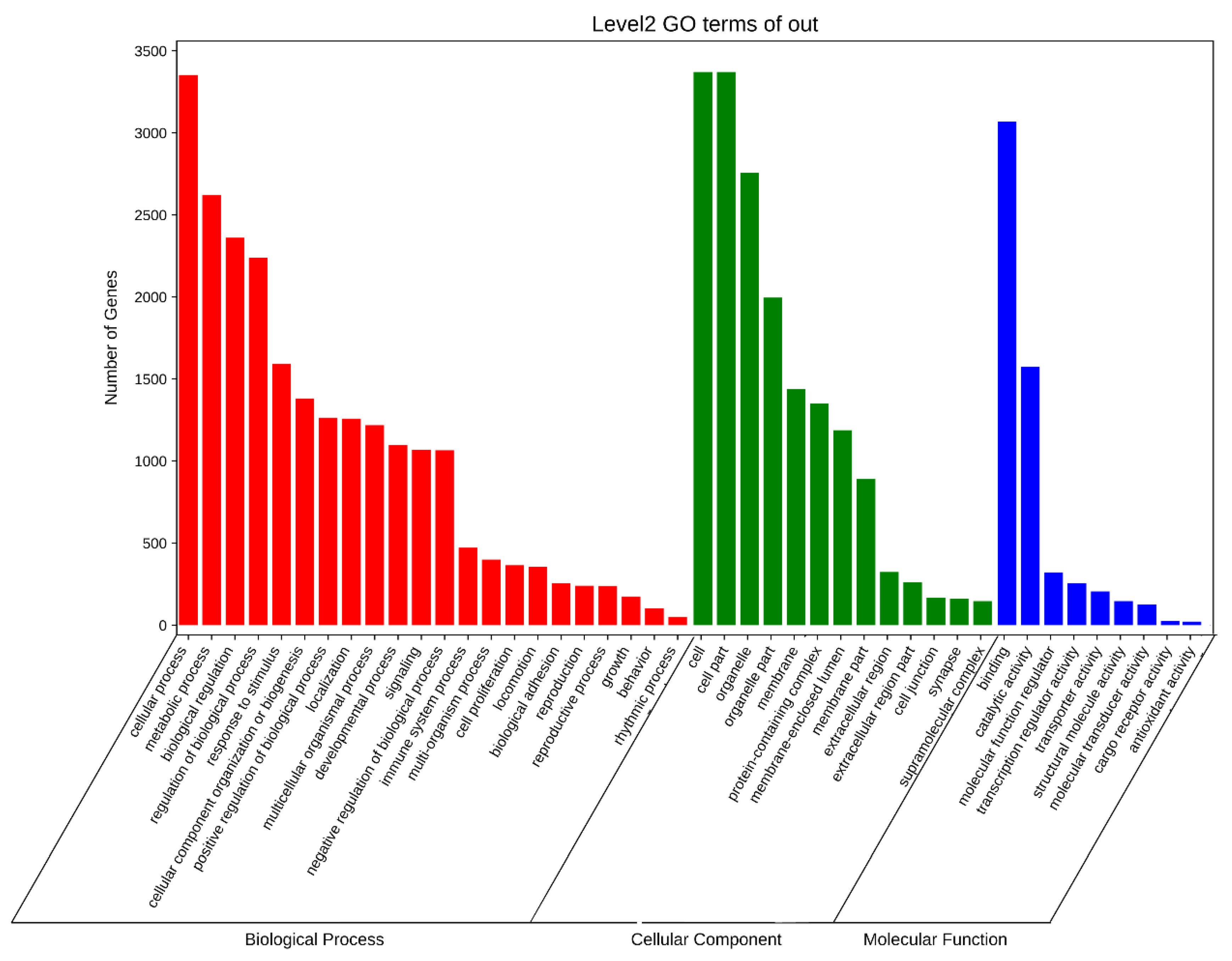
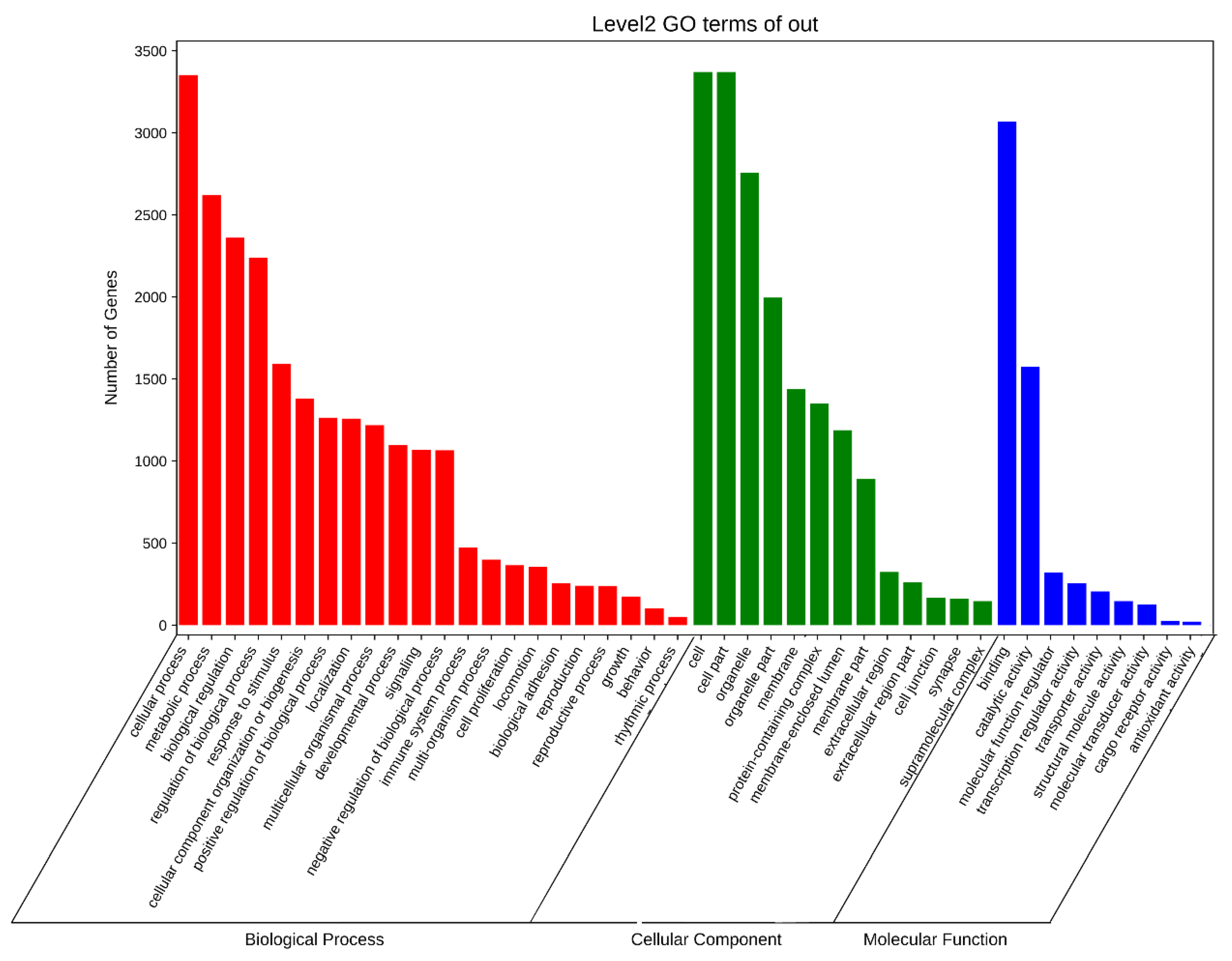
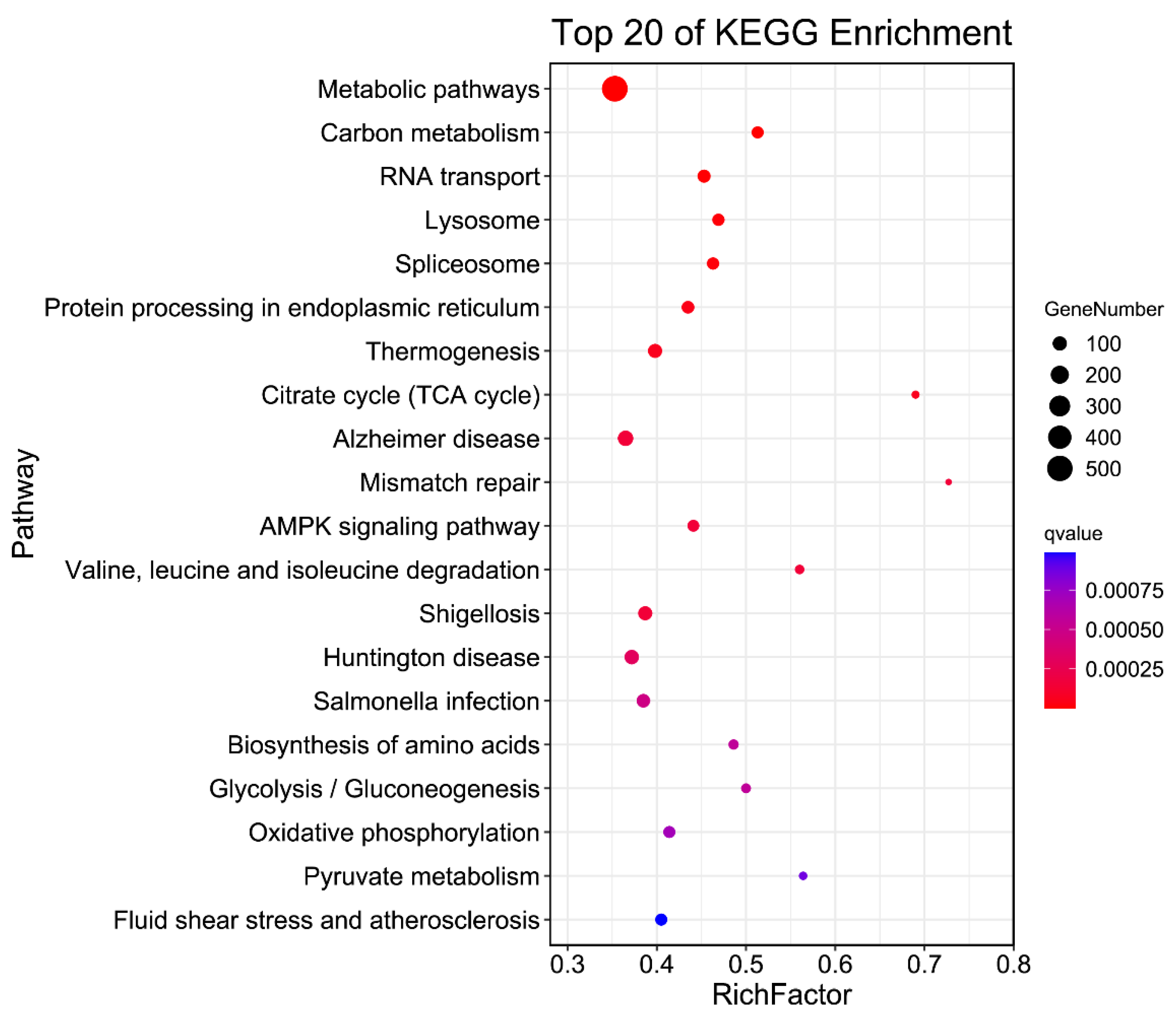
3.6. APA Gene Participates in Diversity Function and Pathway in Pigs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Z.; Li, Y.; Krug, R.M. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, A.W.; Haghighat, A.; Yu, A.T.; Sonenberg, N. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 1998, 392, 520–523. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. A turnover pathway for both stable and unstable mRNAs in yeast: Evidence for a requirement for deadenylation. Genes Dev. 1993, 7, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Edwalds-Gilbert, G.; Veraldi, K.L.; Milcarek, C. Alternative poly(A) site selection in complex transcription units: Means to an end? Nucleic Acids Res. 1997, 25, 2547–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Day, N.; Trifillis, P.; Kiledjian, M. An mRNA Stability Complex Functions with Poly(A)-Binding Protein To Stabilize mRNA In Vitro. Mol. Cell. Biol. 1999, 19, 4552–4560. [Google Scholar] [CrossRef] [Green Version]
- Zarudnaya, M.I.; Hovorun, D.M. Hypothetical double-helical poly(A) formation in a cell and its possible biological significance. IUBMB Life 1999, 48, 581–584. [Google Scholar] [CrossRef]
- Elkon, R.; Ugalde, A.P.; Agami, R. Alternative cleavage and polyadenylation: Extent, regulation and function. Nat. Rev. Genet. 2013, 14, 496–506. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Shenk, T. The sequence 5′-AAUAAA-3′forms parts of the recognition site for polyadenylation of late SV40 mRNAs. Cell 1981, 24, 251–260. [Google Scholar] [CrossRef]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; Macdonald, C.C.; Graber, J.H. Systematic variation in mRNA 3′-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark. Res. 2020, 8, 67. [Google Scholar] [CrossRef]
- Beaudoing, E.; Freier, S.; Wyatt, J.R.; Claverie, J.-M.; Gautheret, D. Patterns of Variant Polyadenylation Signal Usage in Human Genes. Genome Res. 2000, 10, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, C.C.; Redondo, J.L. Reexamining the polyadenylation signal: Were we wrong about AAUAAA? Mol. Cell. Endocrinol. 2002, 190, 1–8. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Retelska, D.; Iseli, C.; Bucher, P.; Jongeneel, C.V.; Naef, F. Similarities and differences of polyadenylation signals in human and fly. BMC Genom. 2006, 7, 176. [Google Scholar] [CrossRef] [Green Version]
- Kamasawa, M.; Horiuchi, J.-I. Identification and characterization of polyadenylation signal (PAS) variants in human genomic sequences based on modified EST clustering. Silico Biol. 2008, 8, 347–361. [Google Scholar]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.J.; Schmidt, R.; Gruber, A.R.; Martin, G.; Ghosh, S.; Belmadani, M.; Keller, W.; Zavolan, M. A comprehensive analysis of 3′ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016, 26, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmanan, V.; Bansal, D.; Kulkarni, J.; Poduval, D.; Krishna, S.; Sasidharan, V.; Anand, P.; Seshasayee, A.; Palakodeti, D. Genome-Wide Analysis of Polyadenylation Events in Schmidtea mediterranea. G3 Genes Genomes Genet. 2016, 6, 3035–3048. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, K.; Sun, Y.; Tong, L. Biophysical characterizations of the recognition of the AAUAAA polyadenylation signal. RNA 2019, 25, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, X.; Ji, G.; Liang, C.; Li, Q.Q. Genome-Wide Comparative Analyses of Polyadenylation Signals in Eukaryotes Suggest a Possible Origin of the AAUAAA Signal. Int. J. Mol. Sci. 2019, 20, 958. [Google Scholar] [CrossRef] [Green Version]
- Higgs, D.R.; Goodbourn, S.E.; Lamb, J.; Clegg, J.B.; Weatherall, D.J.; Proudfoot, N.J. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature 1983, 306, 398–400. [Google Scholar] [CrossRef]
- Bennett, C.L.; Brunkow, M.E.; Ramsdell, F.; O’Briant, K.C.; Zhu, Q.; Fuleihan, R.L.; Shigeoka, A.O.; Ochs, H.D.; Chance, P.F. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 2001, 53, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Shulman, E.D.; Elkon, R. Systematic identification of functional SNPs interrupting 3′UTR polyadenylation signals. PLoS Genet. 2020, 16, e1008977. [Google Scholar] [CrossRef]
- Johnston, J.J.; Williamson, K.A.; Chou, C.M.; Sapp, J.; Ansari, M.; Chapman, H.M.; Cooper, D.N.; Dabir, T.; Dudley, J.N.; Holt, R.J.; et al. NAA10 polyadenylation signal variants cause syndromic microphthalmia. J. Med. Genet. 2019, 56, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Joubert, R.; Mariot, V.; Charpentier, M.; Concordet, J.P.; Dumonceaux, J. Gene editing targeting the DUX4 polyadenylation signal: A Therapy for FSHD? J. Pers. Med. 2020, 11, 7. [Google Scholar] [CrossRef]
- Spöring, M.; Boneberg, R.; Hartig, J.S. Aptamer-Mediated Control of Polyadenylation for Gene Expression Regulation in Mammalian Cells. ACS Synth. Biol. 2020, 9, 3008–3018. [Google Scholar] [CrossRef] [PubMed]
- Derti, A.; Garrett-Engele, P.; MacIsaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 2012, 10, 133–139. [Google Scholar] [CrossRef]
- Neve, J.; Furger, A. Alternative polyadenylation: Less than meets the eye? Biochem. Soc. Trans. 2014, 42, 1190–1195. [Google Scholar] [CrossRef]
- Danckwardt, S.; Hentze, M.W.; Kulozik, A.E. 3′ end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J. 2008, 27, 482–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Ji, G.; Haas, B.J.; Wu, X.; Zheng, J.; Reese, G.J.; Li, Q.Q. Genome level analysis of rice mRNA 3′-end processing signals and alternative polyadenylation. Nucleic Acids Res. 2008, 36, 3150–3161. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, T.; Saito-Takatsuji, H.; Yoshitomi, Y.; Yonekura, H. Role of arginine methylation in alternative polyadenylation of VEGFR-1 (Flt-1) pre-mRNA. Int. J. Mol. Sci. 2020, 21, 6460. [Google Scholar] [CrossRef]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Wang, H.; Li, R.; Zhou, X.; Xue, L.; Xu, X.; Liu, B. Genome-Wide Analysis and Functional Characterization of the Polyadenylation Site in Pigs Using RNAseq Data. Sci. Rep. 2016, 6, 36388. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, J.K.; Marioni, J.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.-B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dai, Z. SANPolyA: A deep learning method for identifying Poly(A) signals. Bioinformatics 2020, 36, 2393–2400. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ye, W.; Ye, L.; Fu, H.; Ye, C.; Xiao, X.; Ji, Y.; Lin, W.; Ji, G.; Wu, X. PlantAPAdb: A Comprehensive Database for Alternative Polyadenylation Sites in Plants. Plant Physiol. 2020, 182, 228–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3’ end sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Types | EST | RNA-Seq |
|---|---|---|
| Raw reads | 1,676,405 | 36,890,214,409 |
| poly(A) tail-containing reads | 105,103 | 352,764,416 |
| poly(A) tail-containing reads uniquely mapped to the genome | 69,602 | 10,938,823 |
| Cleavage sites | 14,845 | 60,277 |
| Total | 62,491 | |
| PAS Motif | Frequency (%) |
|---|---|
| AATAAA | 50.96 |
| ATTAAA | 12.39 |
| TATAAA | 4.45 |
| AGTAAA | 3.93 |
| AAGAAA | 2.63 |
| AATATA | 2.25 |
| AATACA | 2.23 |
| CATAAA | 2.25 |
| GATAAA | 2.09 |
| AATGAA | 0.59 |
| TTTAAA | 1.15 |
| ACTAAA | 1.36 |
| AATAGA | 1.21 |
| AAAAAG | 1.89 |
| None | 10.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Song, J.; Zhang, M.; Deng, Z. Analysis Polyadenylation Signal Usage in Sus scrofa. Animals 2022, 12, 194. https://doi.org/10.3390/ani12020194
Zhang Y, Song J, Zhang M, Deng Z. Analysis Polyadenylation Signal Usage in Sus scrofa. Animals. 2022; 12(2):194. https://doi.org/10.3390/ani12020194
Chicago/Turabian StyleZhang, Yuting, Jingwen Song, Min Zhang, and Zhongyuan Deng. 2022. "Analysis Polyadenylation Signal Usage in Sus scrofa" Animals 12, no. 2: 194. https://doi.org/10.3390/ani12020194
APA StyleZhang, Y., Song, J., Zhang, M., & Deng, Z. (2022). Analysis Polyadenylation Signal Usage in Sus scrofa. Animals, 12(2), 194. https://doi.org/10.3390/ani12020194