



Differential Spleen miRNA Expression Profile of Beagle Dogs Infected with Toxocara canis
 , , and
, , and 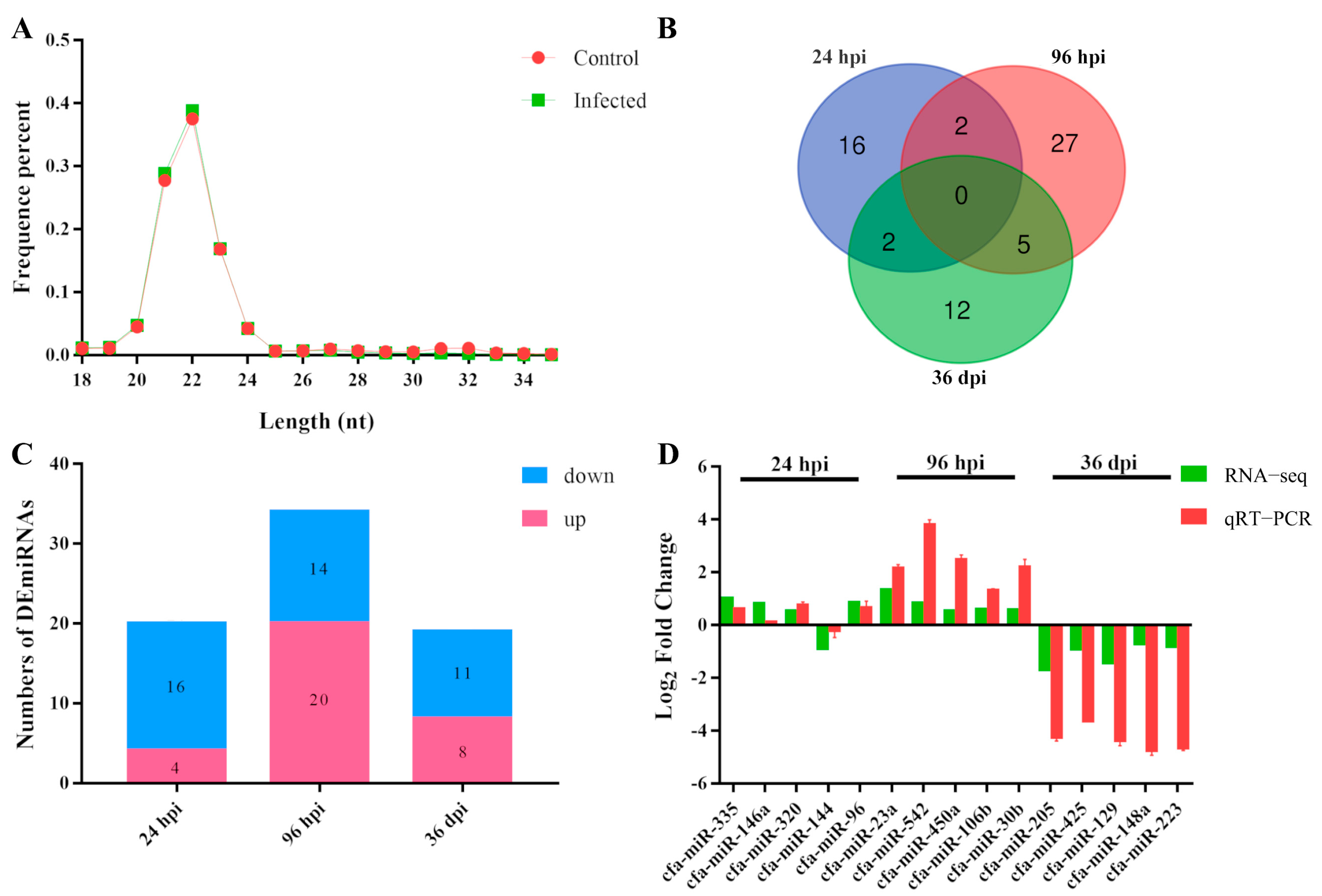{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Animal Infection and Sample Collection
2.3. RNA Extraction and RNA Sequencing
2.4. Identification of miRNAs and Differentially Expressed miRNAs
2.5. GO and KEGG Enrichment Analysis
2.6. Real-Time Quantitative PCR
3. Results
3.1. Characteristic Features of RNA-Sequencing Data
3.2. Patterns of DEmiRNAs
3.3. Quantitative RT-PCR Validation of RNA-Seq Results
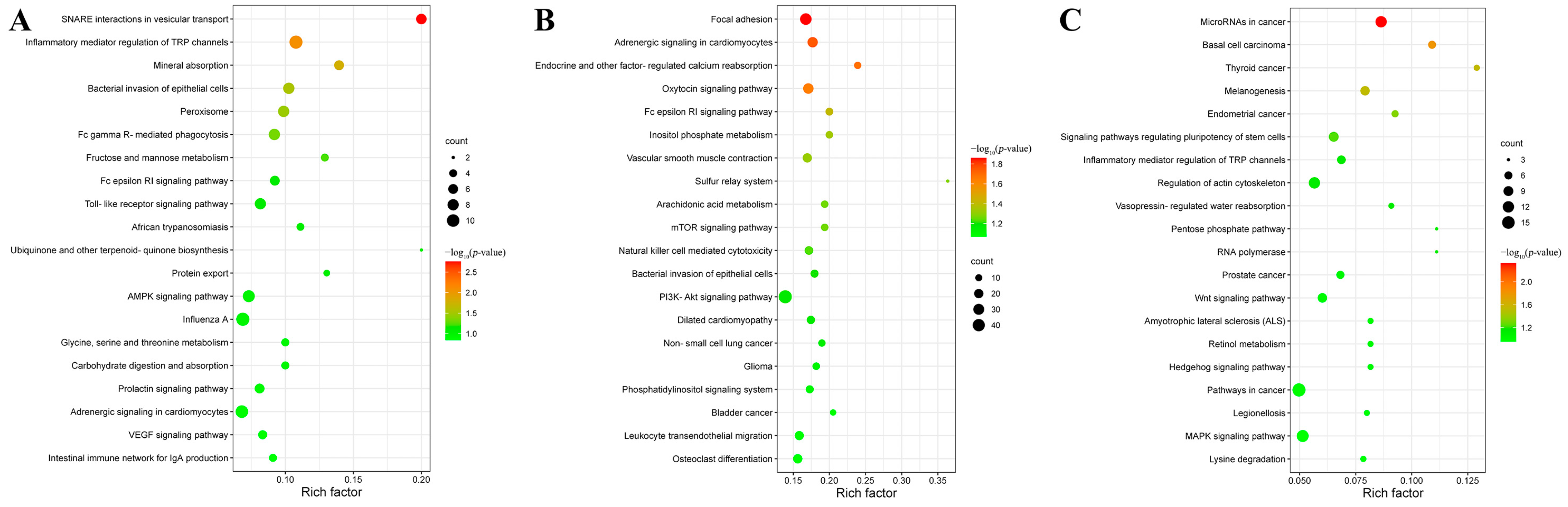
3.4. Target Gene Prediction and Functional Analysis of DEmiRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, C.K.; Holland, C.V.; Loxton, K.; Barghouth, U. Cerebral toxocariasis: Silent progression to neurodegenerative disorders? Clin. Microbiol. Rev. 2015, 28, 663–686. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, Q.; Liu, G.H.; Zheng, W.B.; Hong, S.J.; Sugiyama, H.; Zhu, X.Q.; Elsheikha, H.M. Toxocariasis: A silent threat with a progressive public health impact. Infect. Dis. Poverty 2018, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.B.; Zou, Y.; Liu, Q.; Hu, M.H.; Elsheikha, H.M.; Zhu, X.Q. Toxocara canis infection alters lncRNA and mRNA expression profiles of dog bone marrow. Front. Cell Dev. Biol. 2021, 9, 688128. [Google Scholar] [CrossRef] [PubMed]
- Shepack, A.; Catenazzi, A. Transcriptomics reveal immune downregulation of newts overwhelmed by chytrid co-infection. Mol. Ecol. 2020, 29, 3167–3169. [Google Scholar] [CrossRef]
- Liyanage, T.D.; Nikapitiya, C.; Lee, J.; De Zoysa, M. Molecular insight into regulation of miRNAs in the spleen of zebrafish (Danio rerio) upon pathogenic Streptococcus parauberis infection. Fish Shellfish Immunol. 2020, 106, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef]
- Valencia-Sanchez, M.A.; Liu, J.; Hannon, G.J.; Parker, R. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 2006, 20, 515–524. [Google Scholar] [CrossRef]
- Bensaoud, C.; Hackenberg, M.; Kotsyfakis, M. Noncoding RNAs in parasite-vector-host interactions. Trends Parasitol. 2019, 35, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zheng, W.B.; He, J.J.; Elsheikha, H.M.; Zhu, X.Q.; Lu, Y.X. Toxocara canis differentially affects hepatic microRNA expression in Beagle dogs at different stages of infection. Front. Vet. Sci. 2020, 7, 587273. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Williams, A.; Eisenbarth, S.C. Structure and function of the immune system in the spleen. Sci. Immunol. 2019, 33, eaau6085. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zheng, W.B.; Li, H.Y.; Cai, L.; Zou, Y.; Xie, S.C.; Zhu, X.Q.; Elsheikha, H.M. RNA sequencing reveals dynamic expression of spleen lncRNAs and mRNAs in Beagle dogs infected by Toxocara canis. Parasit. Vectors 2022, 15, 279. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.B.; Zou, Y.; He, J.J.; Elsheikha, H.M.; Liu, G.H.; Hu, M.H.; Wang, S.L.; Zhu, X.Q. Global profiling of lncRNAs-miRNAs-mRNAs reveals differential expression of coding genes and non-coding RNAs in the lung of Beagle dogs at different stages of toxocara canis infection. Int. J. Parasitol. 2021, 51, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. mirdeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids. Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. mirevo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef]
- Zhou, L.; Chen, J.; Li, Z.; Li, X.; Hu, X.; Huang, Y.; Zhao, X.; Liang, C.; Wang, Y.; Sun, L.; et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE 2010, 5, e15224. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Liu, L.; Li, X. Downregulation of miR-320 alleviates endoplasmic reticulum stress and inflammatory response in 3T3-L1 adipocytes. Exp. Clin. Endocrinol. Diabetes 2021, 129, 131–137. [Google Scholar] [CrossRef]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Yin, B.; Umar, T.; Ma, X.; Chen, Y.; Chen, N.; Wu, Z.; Deng, G. miR-193a-3p targets LGR4 to promote the inflammatory response in endometritis. Int. Immunopharmacol. 2021, 98, 107718. [Google Scholar] [CrossRef]
- Foey, A.D.; Feldmann, M.; Brennan, F.M. CD40 ligation induces macrophage IL10 and TNF-α production: Differential use of the PI3K and p42/44 MAPK-pathways. Cytokine 2001, 16, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Fragale, A.; Gabriele, L.; Stellacci, E.; Borghi, P.; Perrotti, E.; Ilari, R.; Lanciotti, A.; Remoli, A.L.; Venditti, M.; Belardelli, F.; et al. IFN regulatory factor-1 negatively regulates CD4+ CD25+ regulatory T cell differentiation by repressing Foxp3 expression. J. Immunol. 2008, 181, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Ogasawara, K.; Takaoka, A.; Tanaka, N. IRF family of transcription factors as regulators of host defense. Annu. Rev. Immunol. 2001, 19, 623–655. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Hu, Y.; Guo, W.; Wang, W.; Yang, Q.; Dong, Q.; Tang, Y.; Li, Q.; Tang, Z. miR-331-3p inhibits inflammatory response after intracerebral hemorrhage by directly targeting NLRP6. Biomed. Res. Int. 2020, 2020, 6182464. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.C.; Alliot, F.; Rutin, J.; Fritz, R.; Trisler, D.; Pessac, B. The myelin basic protein gene is expressed in differentiated blood cell lineages and in hemopoietic progenitors. Proc. Natl. Acad. Sci. USA 2002, 99, 8856–8861. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Chen, B.; Chen, S.; Zhang, M.; Duan, L.; Feng, X.; Chen, J.; Zhou, L.; Chen, L.; Duan, Y. MBP-activated autoimmunity plays a role in arsenic-induced peripheral neuropathy and the potential protective effect of mecobalamin. Environ. Toxicol. 2021, 36, 1243–1253. [Google Scholar] [CrossRef]
- Körner, A.; Bernard, A.; Fitzgerald, J.C.; Alarcon-Barrera, J.C.; Kostidis, S.; Kaussen, T.; Giera, M.; Mirakaj, V. Sema7A is crucial for resolution of severe inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017527118. [Google Scholar] [CrossRef]
- Suzuki, K.; Okuno, T.; Yamamoto, M.; Pasterkamp, R.J.; Takegahara, N.; Takamatsu, H.; Kitao, T.; Takagi, J.; Rennert, P.D.; Kolodkin, A.L.; et al. Semaphorin 7A initiates T-cell-mediated inflammatory responses through α1β1 integrin. Nature 2007, 446, 680–684. [Google Scholar] [CrossRef]
- Greco, E.; Aita, A.; Galozzi, P.; Gava, A.; Sfriso, P.; Negm, O.H.; Tighe, P.; Caso, F.; Navaglia, F.; Dazzo, E.; et al. The novel S59P mutation in the TNFRSF1A gene identified in an adult onset TNF receptor associated periodic syndrome (TRAPS) constitutively activates NF-κB pathway. Arthritis Res. Ther. 2015, 17, 93. [Google Scholar] [CrossRef]
- Hu, Z.; Li, H.; Xie, R.; Wang, S.; Yin, Z.; Liu, Y. Genomic variant in porcine TNFRSF1A gene and its effects on TNF signaling pathway in vitro. Gene 2019, 700, 105–109. [Google Scholar] [CrossRef]
- Kulathu, Y.; Hobeika, E.; Turchinovich, G.; Reth, M. The kinase Syk as an adaptor controlling sustained calcium signalling and B-cell development. EMBO J. 2008, 27, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, J.Y.; Jung, K.I.; Kim, T.J. Characterization of a novel gene in the extended MHC region of mouse, NG29/Cd320, a homolog of the human CD320. Immune Netw. 2009, 9, 138–146. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Zhang, X.; Kovacic, S.; Long, A.J.; Bourque, K.; Wood, C.R.; Choi, Y.S. Identification of a human follicular dendritic cell molecule that stimulates germinal center B cell growth. J. Exp. Med. 2000, 191, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Jung, J.; Xiang, S.; Hollmann, C.; Choi, Y.S. The distinct roles of T cell-derived cytokines and a novel follicular dendritic cell-signaling molecule 8D6 in germinal center-B cell differentiation. J. Immunol. 2001, 167, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Zaccagnini, G.; Greco, S.; Longo, M.; Maimone, B.; Voellenkle, C.; Fuschi, P.; Carrara, M.; Creo, P.; Maselli, D.; Tirone, M.; et al. Hypoxia-induced miR-210 modulates the inflammatory response and fibrosis upon acute ischemia. Cell Death Dis. 2021, 12, 435. [Google Scholar] [CrossRef] [PubMed]
- Virga, F.; Cappellesso, F.; Stijlemans, B.; Henze, A.T.; Trotta, R.; Van Audenaerde, J.; Mirchandani, A.S.; Sanchez-Garcia, M.A.; Vandewalle, J.; Orso, F.; et al. Macrophage miR-210 induction and metabolic reprogramming in response to pathogen interaction boost life-threatening inflammation. Sci. Adv. 2021, 7, eabf0466. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, Z.; Zhang, N.; Song, H.; Zhang, Q.; Hao, X.; Wang, Z. miR-125a-3p inhibits cell proliferation and inflammation responses in fibroblast-like synovial cells in rheumatoid arthritis by mediating the Wnt/β-catenin and NF-κB pathways via targeting MAST3. J. Musculoskelet Neuronal Interact. 2021, 21, 560–567. [Google Scholar] [PubMed]
- Wang, J.K.; Wang, Z.; Li, G. microRNA-125 in immunity and cancer. Cancer Lett. 2019, 454, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.M.; Wu, J.; Zhang, H.; Shi, G.; Chen, Z.T. Circulating miR-125a but not miR-125b is decreased in active disease status and negatively correlates with disease severity as well as inflammatory cytokines in patients with Crohn’s disease. World J. Gastroenterol. 2017, 23, 7888–7898. [Google Scholar] [CrossRef] [PubMed]
- Bemelmans, M.H.A.; Van Tits, L.J.H.; Buurman, W.A. Tumor necrosis factor: Function, release and clearance. Crit. Rev. Immunol. 2017, 37, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Varfolomeev, E.; Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 2018, 101, 26–32. [Google Scholar] [CrossRef]
- Luo, Z.F.; Jiang, X.H.; Liu, H.; He, L.Y.; Luo, X.; Chen, F.C.; Tan, Y.L. miR-23b attenuates LPS-induced inflammatory responses in acute lung injury via inhibition of HDAC2. Biochem. Genet. 2021, 59, 604–616. [Google Scholar] [CrossRef]
- Zambrano, S.; Möller-Hackbarth, K.; Li, X.; Rodriguez, P.Q.; Charrin, E.; Schwarz, A.; Nyström, J.; Wernerson, A.Ö.; Lal, M.; Patrakka, J. GPRC5b modulates inflammatory response in glomerular diseases via NF-κB pathway. J. Am. Soc. Nephrol. 2019, 30, 1573–1586. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, G.; Di, Z.; Zhao, Q. miR-339-5p inhibits alcohol-induced brain inflammation through regulating NF-κB pathway. Biochem. Biophys. Res. Commun. 2014, 452, 450–456. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Kuroda, E.; Yoshida, Y.; En Shan, B.; Yamashita, U. Suppression of macrophage interleukin-12 and tumour necrosis factor-alpha production in mice infected with Toxocara canis. Parasite Immunol. 2001, 23, 305–311. [Google Scholar] [CrossRef]
- Duan, X.; Zohaib, A.; Li, Y.; Zhu, B.; Ye, J.; Wan, S.; Xu, Q.; Song, Y.; Chen, H.; Cao, S. miR-206 modulates lipopolysaccharide-mediated inflammatory cytokine production in human astrocytes. Cell. Signal. 2015, 27, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Deng, Y.; Liu, L.; Yu, K.; Zhang, L.; Wang, H.; He, X.; Wang, J.; Lu, C.; Wu, L.N.; et al. Dual regulatory switch through interactions of tcf7l2/tcf4 with stage-specific partners propels oligodendroglial maturation. Nat. Commun. 2016, 7, 10883. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Spitzner, M.; Reineke, S.; Möller, J.; Auslander, N.; Kramer, F.; Hu, Y.; Beissbarth, T.; Wolff, H.A.; Rave-Fränk, M.; et al. Chemoradiotherapy resistance in colorectal cancer cells is mediated by Wnt/β-catenin signaling. Mol. Cancer Res. 2017, 15, 1481–1490. [Google Scholar] [CrossRef]
- Hrckulak, D.; Kolar, M.; Strnad, H.; Korinek, V. TCF/LEF transcription factors: An update from the internet resources. Cancers 2016, 8, 70. [Google Scholar] [CrossRef]
- Gao, Z.; Li, Q.; Zhang, Y.; Gao, X.; Li, H.; Yuan, Z. Ripasudil alleviated the inflammation of RPE cells by targeting the miR-136-5p/ROCK/NLRP3 pathway. BMC Ophthalmol. 2020, 20, 134. [Google Scholar] [CrossRef]
- Wright, J.F.; Bennett, F.; Li, B.; Brooks, J.; Luxenberg, D.P.; Whitters, M.J.; Tomkinson, K.N.; Fitz, L.J.; Wolfman, N.M.; Collins, M.; et al. The human L-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J. Immunol. 2008, 181, 2799–2805. [Google Scholar] [CrossRef]
- Puel, A.; Cypowyj, S.; Maródi, L.; Abel, L.; Picard, C.; Casanova, J.L. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 616–622. [Google Scholar] [CrossRef]
- Kuestner, R.E.; Taft, D.W.; Haran, A.; Brandt, C.S.; Brender, T.; Lum, K.; Harder, B.; Okada, S.; Ostrander, C.D.; Kreindler, J.L.; et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J. Immunol. 2007, 179, 5462–5473. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.P.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Li, H.-Y.; Cai, L.; Xie, S.-C.; Zou, Y.; Zhu, X.-Q.; Zheng, W.-B. Differential Spleen miRNA Expression Profile of Beagle Dogs Infected with Toxocara canis. Animals 2022, 12, 2638. https://doi.org/10.3390/ani12192638
Xu Y, Li H-Y, Cai L, Xie S-C, Zou Y, Zhu X-Q, Zheng W-B. Differential Spleen miRNA Expression Profile of Beagle Dogs Infected with Toxocara canis. Animals. 2022; 12(19):2638. https://doi.org/10.3390/ani12192638
Chicago/Turabian StyleXu, Yue, Hao-Yu Li, Lang Cai, Shi-Chen Xie, Yang Zou, Xing-Quan Zhu, and Wen-Bin Zheng. 2022. "Differential Spleen miRNA Expression Profile of Beagle Dogs Infected with Toxocara canis" Animals 12, no. 19: 2638. https://doi.org/10.3390/ani12192638
APA StyleXu, Y., Li, H.-Y., Cai, L., Xie, S.-C., Zou, Y., Zhu, X.-Q., & Zheng, W.-B. (2022). Differential Spleen miRNA Expression Profile of Beagle Dogs Infected with Toxocara canis. Animals, 12(19), 2638. https://doi.org/10.3390/ani12192638