
Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Published ASFV Genome Sequences Acquisition
2.2. Sequence Alignment
2.3. Maximum Likelihood Phylogenetic Analysis
2.4. Sequence Diversity Analysis
2.5. ORF Analysis
2.6. Tandem Repeat Sequences Analysis
3. Results
3.1. Curated ASFV Complete Genome Sequences Dataset
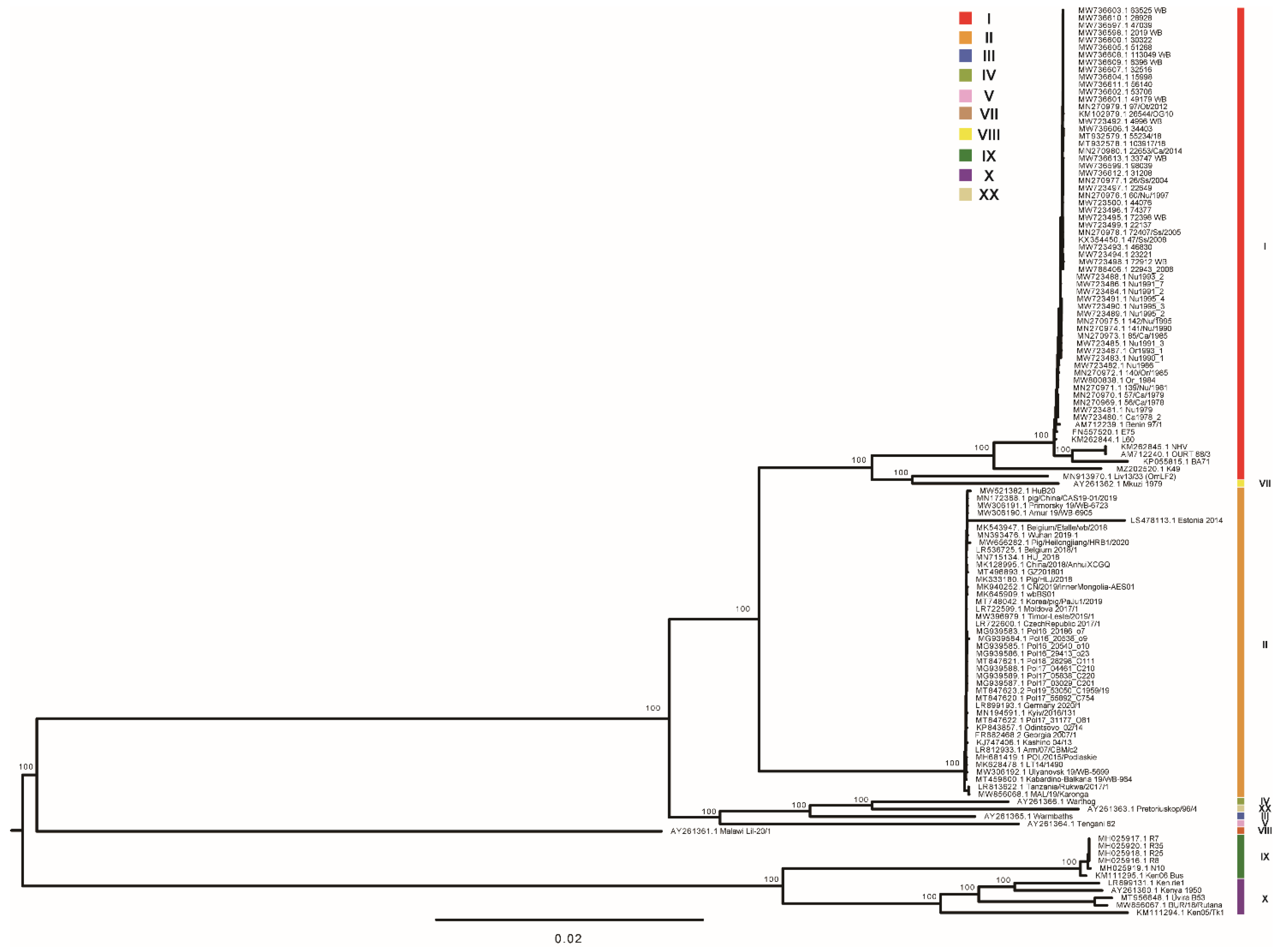
3.2. Phylogenetic Reconstruction
3.3. Genome-Wide Sequence Similarity
3.4. Genome Annotation
3.5. Structural Variation
3.6. Tandem Repeat Sequences Variation
3.7. Highly Variable Poly G or Poly C Tracts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, L.K.; Sun, H.; Roberts, H. African swine fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Penrith, M.L.; Kivaria, F.M. One hundred years of African swine fever in Africa: Where have we been, where are we now, where are we going? Transbound. Emerg. Dis. 2022. [Google Scholar] [CrossRef] [PubMed]
- OIE, African Swine Fever. 2022. Available online: https://www.woah.org/en/disease/african-swine-fever/#ui-id-2 (accessed on 10 November 2021).
- Woonwong, Y.; Do Tien, D.; Thanawongnuwech, R. The Future of the Pig Industry After the Introduction of African Swine Fever into Asia. Anim. Front. Rev. Mag. Anim. Agric. 2020, 10, 30–37. [Google Scholar] [CrossRef]
- McCarthy, C.P.; Vaduganathan, M.; Solomon, E.; Sakhuja, R.; Piazza, G.; Bhatt, D.L.; Connors, J.M.; Patel, N.K. Running thin: Implications of a heparin shortage. Lancet 2020, 395, 534–536. [Google Scholar] [CrossRef]
- Schulz, K.; Boklund, A. The Epidemiology of African Swine Fever, Its Complexity and the Requirement for Multiple Solution Approaches. Animals 2020, 10, 1900. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Costard, S.; Mur, L.; Lubroth, J.; Sanchez-Vizcaino, J.M.; Pfeiffer, D.U. Epidemiology of African swine fever virus. Virus Res. 2013, 173, 191–197. [Google Scholar] [CrossRef]
- Normile, D. African swine fever marches across much of Asia. Science 2019, 364, 617–618. [Google Scholar] [CrossRef]
- Stokstad, E. Deadly virus threatens European pigs and boar. Science 2017, 358, 1516–1517. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernandez-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernandez-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Nix, R.J.; Gallardo, C.; Hutchings, G.; Blanco, E.; Dixon, L.K. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch. Virol. 2006, 151, 2475–2494. [Google Scholar] [CrossRef]
- Malogolovkin, A.; Yelsukova, A.; Gallardo, C.; Tsybanov, S.; Kolbasov, D. Molecular characterization of African swine fever virus isolates originating from outbreaks in the Russian Federation between 2007 and 2011. Vet. Microbiol. 2012, 158, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; King, J.; Groza, O.; Hubner, A.; Olesen, A.S.; Hoper, D.; Dixon, L.K.; Netherton, C.L.; Rasmussen, T.B.; et al. A Deep-Sequencing Workflow for the Fast and Efficient Generation of High-Quality African Swine Fever Virus Whole-Genome Sequences. Viruses 2019, 11, 846. [Google Scholar] [CrossRef]
- Jia, L.; Jiang, M.; Wu, K.; Hu, J.; Wang, Y.; Quan, W.; Hao, M.; Liu, H.; Wei, H.; Fan, W.; et al. Nanopore sequencing of African swine fever virus. Sci. China Life Sci. 2020, 63, 160–164. [Google Scholar] [CrossRef]
- Forth, J.H.; Tignon, M.; Cay, A.B.; Forth, L.F.; Hoper, D.; Blome, S.; Beer, M. Comparative Analysis of Whole-Genome Sequence of African Swine Fever Virus Belgium 2018/1. Emerg. Infect. Dis. 2019, 25, 1249–1252. [Google Scholar] [CrossRef]
- Bao, J.; Wang, Q.; Lin, P.; Liu, C.; Li, L.; Wu, X.; Chi, T.; Xu, T.; Ge, S.; Liu, Y.; et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 Genotype II strains. Transbound. Emerg. Dis. 2019, 66, 1167–1176. [Google Scholar] [CrossRef]
- Masembe, C.; Sreenu, V.B.; Da Silva Filipe, A.; Wilkie, G.S.; Ogweng, P.; Mayega, F.J.; Muwanika, V.B.; Biek, R.; Palmarini, M.; Davison, A.J. Genome Sequences of Five African Swine Fever Virus Genotype IX Isolates from Domestic Pigs in Uganda. Microbiol. Resour. Announc. 2018, 7, e01018-18. [Google Scholar] [CrossRef]
- Chapman, D.A.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef]
- Chapman, D.A.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89 Pt 2, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Fiori, M.S.; Sanna, D.; Scarpa, F.; Floris, M.; Di Nardo, A.; Ferretti, L.; Loi, F.; Cappai, S.; Sechi, A.M.; Angioi, P.P.; et al. A Deeper Insight into Evolutionary Patterns and Phylogenetic History of ASFV Epidemics in Sardinia (Italy) through Extensive Genomic Sequencing. Viruses 2021, 13, 1994. [Google Scholar] [CrossRef] [PubMed]
- Torresi, C.; Fiori, M.; Bertolotti, L.; Floris, M.; Colitti, B.; Giammarioli, M.; Dei Giudici, S.; Oggiano, A.; Malmberg, M.; De Mia, G.M.; et al. The evolution of African swine fever virus in Sardinia (1978–2014) as revealed by whole-genome sequencing and comparative analysis. Transbound. Emerg. Dis. 2020, 67, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.J.; Jia, H.; Xie, C.D.; Shagainar, J.; Feng, Z.; Zhang, X.; Li, K.; Zhou, R. Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses 2022, 14, 889. [Google Scholar] [CrossRef] [PubMed]
- Forth, J.H.; Forth, L.F.; Blome, S.; Hoper, D.; Beer, M. African swine fever whole-genome sequencing-Quantity wanted but quality needed. PLoS Pathog. 2020, 16, e1008779. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Perez-Nunez, D.; Castillo-Rosa, E.; Vigara-Astillero, G.; Garcia-Belmonte, R.; Gallardo, C.; Revilla, Y. Identification and Isolation of Two Different Subpopulations Within African Swine Fever Virus Arm/07 Stock. Vaccines 2020, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Portugal, R.; Coelho, J.; Hoper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitao, A.; Keil, G.M. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2015, 96 Pt 2, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.T.; Truong, A.D.; Dang, H.V. Whole Genome Sequencing of African Swine Fever. Methods Mol. Biol. 2022, 2503, 205–215. [Google Scholar]
- Olasz, F.; Meszaros, I.; Marton, S.; Kajan, G.L.; Tamas, V.; Locsmandi, G.; Magyar, T.; Balint, A.; Banyai, K.; Zadori, Z. A Simple Method for Sample Preparation to Facilitate Efficient Whole-Genome Sequencing of African Swine Fever Virus. Viruses 2019, 11, 1129. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.P.; Gallardo, C.; Arias, M.; da Silva, M.; Upton, C.; Martin, R.; Bishop, R.P. Phylogenomic analysis of 11 complete African swine fever virus genome sequences. Virology 2010, 400, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.D.; Penrith, M.L.; Cruciere, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Lubisi, B.A.; Bastos, A.D.; Dwarka, R.M.; Vosloo, W. Molecular epidemiology of African swine fever in East Africa. Arch. Virol. 2005, 150, 2439–2452. [Google Scholar] [CrossRef]
- Peter, E.; Machuka, E.; Githae, D.; Okoth, E.; Cleaveland, S.; Shirima, G.; Kusiluka, L.; Pelle, R. Detection of African swine fever virus genotype XV in a sylvatic cycle in Saadani National Park, Tanzania. Transbound. Emerg. Dis. 2020, 68, 813–823. [Google Scholar] [CrossRef]
- Phologane, S.B.; Bastos, A.D.; Penrith, M.L. Intra- and inter-genotypic size variation in the central variable region of the 9RL open reading frame of diverse African swine fever viruses. Virus Genes 2005, 31, 357–360. [Google Scholar] [CrossRef]
- Irusta, P.M.; Borca, M.V.; Kutish, G.F.; Lu, Z.; Caler, E.; Carrillo, C.; Rock, D.L. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology 1996, 220, 20–27. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martin, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. [Google Scholar] [CrossRef] [PubMed]
- Esposito, J.J.; Sammons, S.A.; Frace, A.M.; Osborne, J.D.; Olsen-Rasmussen, M.; Zhang, M.; Govil, D.; Damon, I.K.; Kline, R.; Laker, M.; et al. Genome sequence diversity and clues to the evolution of variola (smallpox) virus. Science 2006, 313, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, S.; Williamson, A.L.; Malesa, R.; van Heerden, J.; Boshoff, C.I.; Bastos, A.D.S.; Heath, L.; Carulei, O. Genome Sequences of Three African Swine Fever Viruses of Genotypes I, III, and XXII from South Africa and Zambia, Isolated from Ornithodoros Soft Ticks. Microbiol. Resour. Announc. 2020, 9, e01376-19. [Google Scholar] [CrossRef] [PubMed]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvili, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef]
- Wen, X.; He, X.; Zhang, X.; Zhang, X.; Liu, L.; Guan, Y.; Zhang, Y.; Bu, Z. Genome sequences derived from pig and dried blood pig feed samples provide important insights into the transmission of African swine fever virus in China in 2018. Emerg. Microbes Infect. 2019, 8, 303–306. [Google Scholar] [CrossRef]
- Sun, E.; Zhang, Z.; Wang, Z.; He, X.; Zhang, X.; Wang, L.; Wang, W.; Huang, L.; Xi, F.; Huangfu, H.; et al. Emergence and prevalence of naturally occurring lower virulent African swine fever viruses in domestic pigs in China in 2020. Sci. China Life Sci. 2021, 64, 752–765. [Google Scholar] [CrossRef]
- Franzoni, G.; Dei Giudici, S.; Loi, F.; Sanna, D.; Floris, M.; Fiori, M.; Sanna, M.L.; Madrau, P.; Scarpa, F.; Zinellu, S.; et al. African Swine Fever Circulation among Free-Ranging Pigs in Sardinia: Data from the Eradication Program. Vaccines 2020, 8, 549. [Google Scholar] [CrossRef]
- Granberg, F.; Torresi, C.; Oggiano, A.; Malmberg, M.; Iscaro, C.; De Mia, G.M.; Belak, S. Complete Genome Sequence of an African Swine Fever Virus Isolate from Sardinia, Italy. Genome Announc. 2016, 4, e01220-16. [Google Scholar] [CrossRef]
- Gilliaux, G.; Garigliany, M.; Licoppe, A.; Paternostre, J.; Lesenfants, C.; Linden, A.; Desmecht, D. Newly emerged African swine fever virus strain Belgium/Etalle/wb/2018: Complete genomic sequence and comparative analysis with reference p72 genotype II strains. Transbound. Emerg. Dis. 2019, 66, 2566–2591. [Google Scholar] [CrossRef]
- Jia, L.; Chen, J.; Liu, H.; Fan, W.; Wang, D.; Li, J.; Liu, D. Potential m6A and m5C Methylations within the Genome of A Chinese African Swine Fever Virus Strain. Virol. Sin. 2020, 36, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Hoper, D.; et al. Deletion at the 5’-end of Estonian ASFV strains associated with an attenuated phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef]
- Gallardo, C.; Soler, A.; Nieto, R.; Cano, C.; Pelayo, V.; Sanchez, M.A.; Pridotkas, G.; Fernandez-Pinero, J.; Briones, V.; Arias, M. Experimental Infection of Domestic Pigs with African Swine Fever Virus Lithuania 2014 Genotype II Field Isolate. Transbound. Emerg. Dis. 2017, 64, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Hakizimana, J.N.; Ntirandekura, J.B.; Yona, C.; Nyabongo, L.; Kamwendo, G.; Chulu, J.L.C.; Ntakirutimana, D.; Kamana, O.; Nauwynck, H.; Misinzo, G. Complete genome analysis of African swine fever virus responsible for outbreaks in domestic pigs in 2018 in Burundi and 2019 in Malawi. Trop. Anim. Health Prod. 2021, 53, 438. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Wozniakowski, G.; Niemczuk, K. The first complete genomic sequences of African swine fever virus isolated in Poland. Sci. Rep. 2019, 9, 4556. [Google Scholar] [CrossRef]
- Olesen, A.S.; Lohse, L.; Dalgaard, M.D.; Wozniakowski, G.; Belsham, G.J.; Botner, A.; Rasmussen, T.B. Complete genome sequence of an African swine fever virus (ASFV POL/2015/Podlaskie) determined directly from pig erythrocyte-associated nucleic acid. J. Virol. Methods 2018, 261, 14–16. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Walczak, M.; Juszkiewicz, M.; Wozniakowski, G. The Spillover of African Swine Fever in Western Poland Revealed Its Estimated Origin on the Basis of O174L, K145R, MGF 505-5R and IGR I73R/I329L Genomic Sequences. Viruses 2020, 12, 1094. [Google Scholar] [CrossRef]
- Mazloum, A.; van Schalkwyk, A.; Shotin, A.; Igolkin, A.; Shevchenko, I.; Gruzdev, K.N.; Vlasova, N. Comparative Analysis of Full Genome Sequences of African Swine Fever Virus Isolates Taken from Wild Boars in Russia in 2019. Pathogens 2021, 10, 521. [Google Scholar] [CrossRef]
- Njau, E.P.; Domelevo Entfellner, J.B.; Machuka, E.M.; Bochere, E.N.; Cleaveland, S.; Shirima, G.M.; Kusiluka, L.J.; Upton, C.; Bishop, R.P.; Pelle, R.; et al. The first genotype II African swine fever virus isolated in Africa provides insight into the current Eurasian pandemic. Sci. Rep. 2021, 11, 13081. [Google Scholar] [CrossRef]
- Mileto, P.; da Conceicao, F.; Stevens, V.; Cummins, D.; Certoma, A.; Neave, M.J.; Bendita da Costa Jong, J.; Williams, D.T. Complete Genome Sequence of African Swine Fever Virus Isolated from a Domestic Pig in Timor-Leste, 2019. Microbiol. Resour. Announc. 2021, 10, e0026321. [Google Scholar] [CrossRef]
- Kovalenko, G.; Ducluzeau, A.L.; Ishchenko, L.; Sushko, M.; Sapachova, M.; Rudova, N.; Solodiankin, O.; Gerilovych, A.; Dagdag, R.; Redlinger, M.; et al. Complete Genome Sequence of a Virulent African Swine Fever Virus from a Domestic Pig in Ukraine. Microbiol. Resour. Announc. 2019, 8, e00883-19. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.P.; Fleischauer, C.; de Villiers, E.P.; Okoth, E.A.; Arias, M.; Gallardo, C.; Upton, C. Comparative analysis of the complete genome sequences of Kenyan African swine fever virus isolates within p72 genotypes IX and X. Virus Genes 2015, 50, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Chastagner, A.; Pereira de Oliveira, R.; Hutet, E.; Le Dimna, M.; Paboeuf, F.; Lucas, P.; Blanchard, Y.; Dixon, L.; Vial, L.; Le Potier, M.F. Coding-Complete Genome Sequence of an African Swine Fever Virus Strain Liv13/33 Isolate from Experimental Transmission between Pigs and Ornithodoros moubata Ticks. Microbiol. Resour. Announc. 2020, 9, e00185-20. [Google Scholar] [CrossRef] [PubMed]
- Bisimwa, P.N.; Ongus, J.R.; Steinaa, L.; Bisimwa, E.B.; Bochere, E.; Machuka, E.M.; Entfellner, J.D.; Okoth, E.; Pelle, R. The first complete genome sequence of the African swine fever virus genotype X and serogroup 7 isolated in domestic pigs from the Democratic Republic of Congo. Virol. J. 2021, 18, 23. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | LVR | RVR | ||||||
|---|---|---|---|---|---|---|---|---|
| MGF 100 | MGF 110 | MGF 300 | MGF 360 | MGF 505/530 | MGF 100 | MGF 360 | MGF 505/530 | |
| I (KM262844.1) | 0 | 6 | 3 | 13 | 9 | 2 | 3 | 1 |
| II (FR682468.2) | 1 | 11 | 3 | 14 | 9 | 2 | 5 | 1 |
| III (AY261365.1) | 1 | 11 | 3 | 13 | 9 | 2 | 4 | 1 |
| IV (AY261366.1) | 1 | 9 | 3 | 11 | 9 | 2 | 4 | 1 |
| V (AY261364.1) | 1 | 6 | 3 | 12 | 9 | 2 | 4 | 1 |
| VII (AY261362.1) | 1 | 10 | 3 | 13 | 9 | 2 | 4 | 1 |
| VIII (AY261361.1) | 1 | 10 | 3 | 12 | 8 | 2 | 3 | 1 |
| IX (KM111295.1) | 1 | 9 | 3 | 10 | 8 | 2 | 3 | 1 |
| X (AY261360.1) | 1 | 11 | 3 | 14 | 8 | 2 | 3 | 1 |
| XX (AY261363.1) | 1 | 9 | 3 | 13 | 9 | 2 | 4 | 1 |
| TRS | Site | Start | Stop | Pattern | RU | Sequence | Length | Number |
|---|---|---|---|---|---|---|---|---|
| TRS1 | NCR MGF505-9R_10R | 45,221 | 45,491 | (A)n | A | GTTCAGTTAAGACAGTA/GTTAAGACAATAGTTTT | 17 | 7–32 |
| TRS2 | EP402R | 75,257 | 75,322 | (A)n | A | AAACCATGTCCTCCACCC | 18 | 7–14 |
| TRS3 | C84L | 82,690 | 82,785 | (A)n | A | GTGCCTGCACAA | 12 | 3–8 |
| TRS4 | NCR C84L_C717R | 82,976 | 83,053 | (A)n | A | GTTTTAGCTT * | 10 | 2–8 |
| TRS5 | NCR C122R_C257L | 85,722 | 85,898 | (A)n-X-(B)n | A | CAAGTATTTTCTATAGC | 17 | 3–7 |
| B | CATTAAAACAAAGAGTCTAATAAGACGCTTTAATGGG * | 37 | 1–6 | |||||
| TRS6 | NCR C315R_C147L | 89,123 | 89,187 | (A)n | A | CTAAACGTTTAAA * | 13 | 3–6 |
| TRS7 | B475L | 99,197 | 99,397 | (A)n-(B)n | A | CAAGGATATTTTCTTTGATAT | 21 | 0–4 |
| B | CAAGAATATTTTCTTCTTCAGGTTTATCCTGACCAAACT * | 39 | 3–5 | |||||
| TRS8 | B602L | 102,670 | 102,789 | (A)n | A | GTGCTTGTACAA | 12 | 8–31 |
| TRS9 | B407L | 108,045 | 108,239 | (A)n | A | CCTCGGCGCGTCTTA | 15 | 10–14 |
| TRS10 | E183L | 163,354 | 163,485 | (A)n-X-(B)n | A | GCGGCCGCAGGT | 12 | 0–7 |
| B | GGGTTGTCCGTAACT | 15 | 3–6 | |||||
| TRS11 | NCR E146L_E199L | 166,659 | 166,816 | (A)n | A | CTTAAGTTTATTGCTCATGG * | 20 | 2–27 |
| TRS12 | NCR I73R_I329L | 173,344 | 173,411 | (A)n-(B)n | A | TAAATGTAGAATAACACAGTTAAGCAATAAATAACAAG | 38 | 1–8 |
| B | TATATAGGAA | 10 | 3–17 | |||||
| TRS13 | NCR I329L_I215L | 174,667 | 174,878 | (A)n-X-(B)n-X-(C)n | A | CTAAATTCTAAGCA | 14 | 3–21 |
| B | CAT | 3 | 11–20 | |||||
| C | TCTTCA | 5 | 3–5 | |||||
| TRS14 | I196L | 176,397 | 176,528 | (A)n | A | TTGTTGTTGTGGGTGAAATAGTTGTACTGGTATTATTGGAAATGGCTGTCATTAAATAATTACTAC * | 66 | 1–3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, J.; Zhang, Y.; Shi, C.; Wang, Q.; Wang, S.; Wu, X.; Cao, S.; Xu, F.; Wang, Z. Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals 2022, 12, 2446. https://doi.org/10.3390/ani12182446
Bao J, Zhang Y, Shi C, Wang Q, Wang S, Wu X, Cao S, Xu F, Wang Z. Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals. 2022; 12(18):2446. https://doi.org/10.3390/ani12182446
Chicago/Turabian StyleBao, Jingyue, Yong Zhang, Chuan Shi, Qinghua Wang, Shujuan Wang, Xiaodong Wu, Shengbo Cao, Fengping Xu, and Zhiliang Wang. 2022. "Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset" Animals 12, no. 18: 2446. https://doi.org/10.3390/ani12182446
APA StyleBao, J., Zhang, Y., Shi, C., Wang, Q., Wang, S., Wu, X., Cao, S., Xu, F., & Wang, Z. (2022). Genome-Wide Diversity Analysis of African Swine Fever Virus Based on a Curated Dataset. Animals, 12(18), 2446. https://doi.org/10.3390/ani12182446