
Adaptability and Evolution of Gobiidae: A Genetic Exploration

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Assembly and Annotation of the Complete Mitogenome
2.2. Phylogenomic Analyses
2.3. Selection Analyses
3. Results
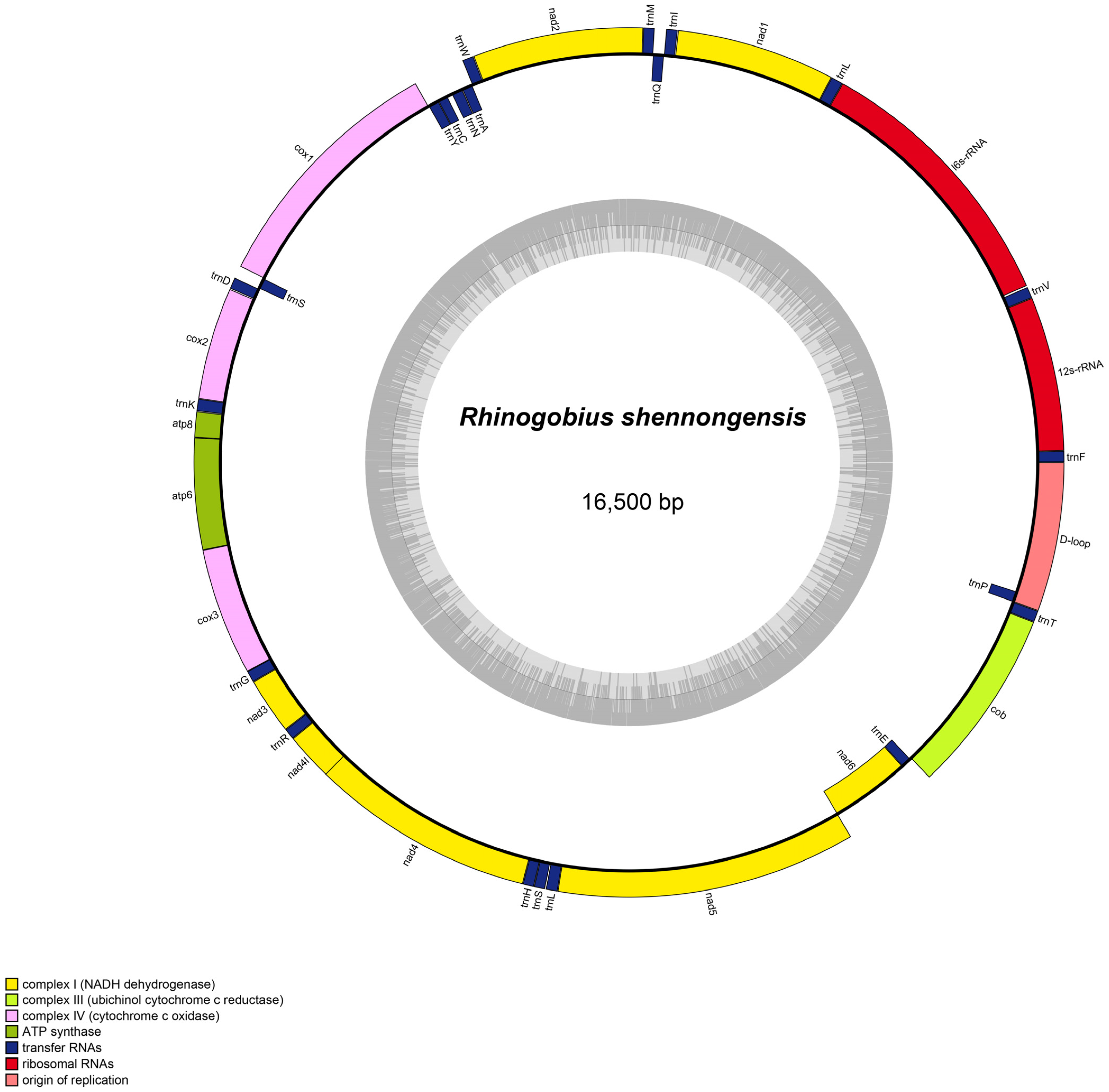
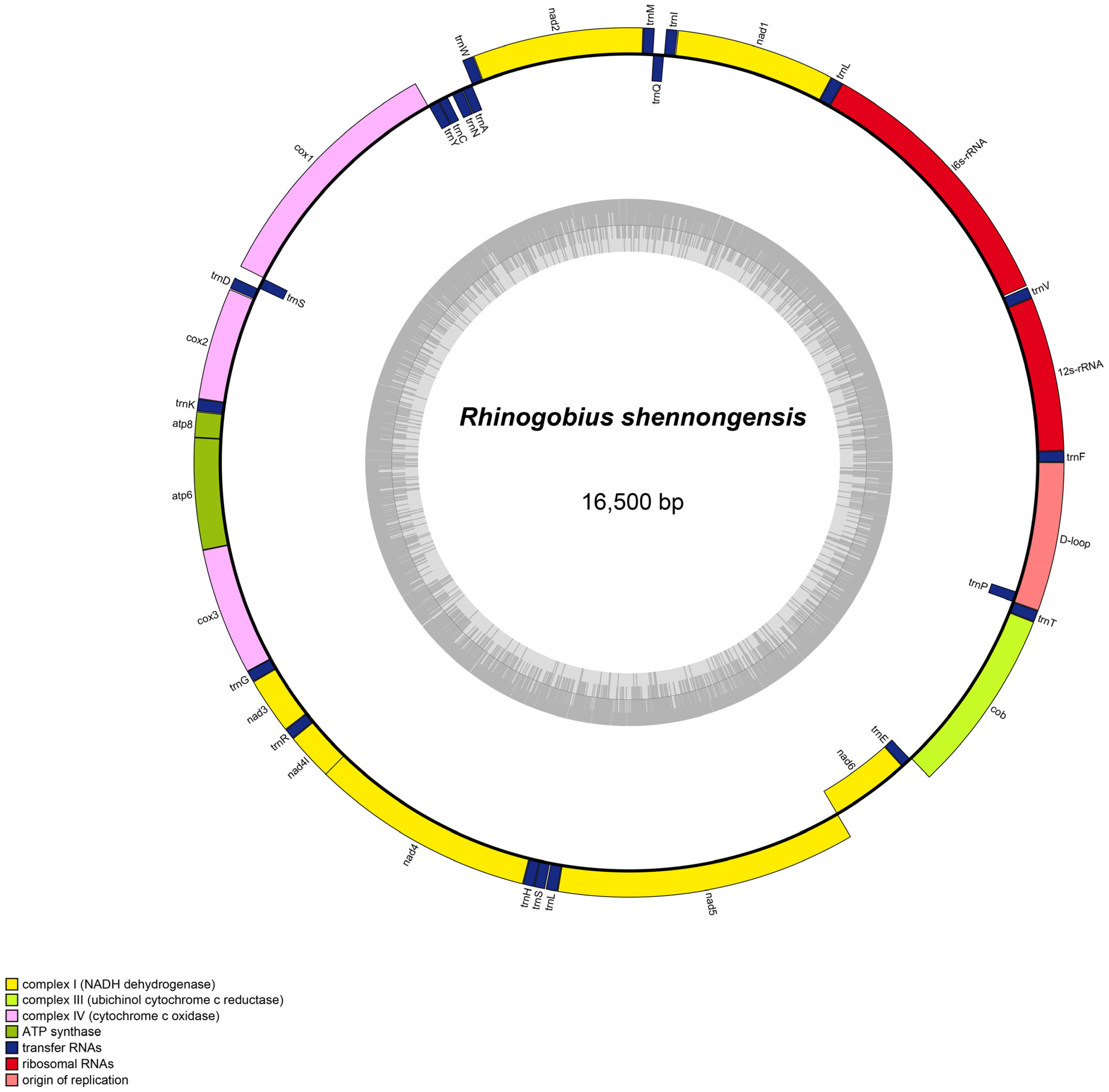
3.1. Mitogenome Organization
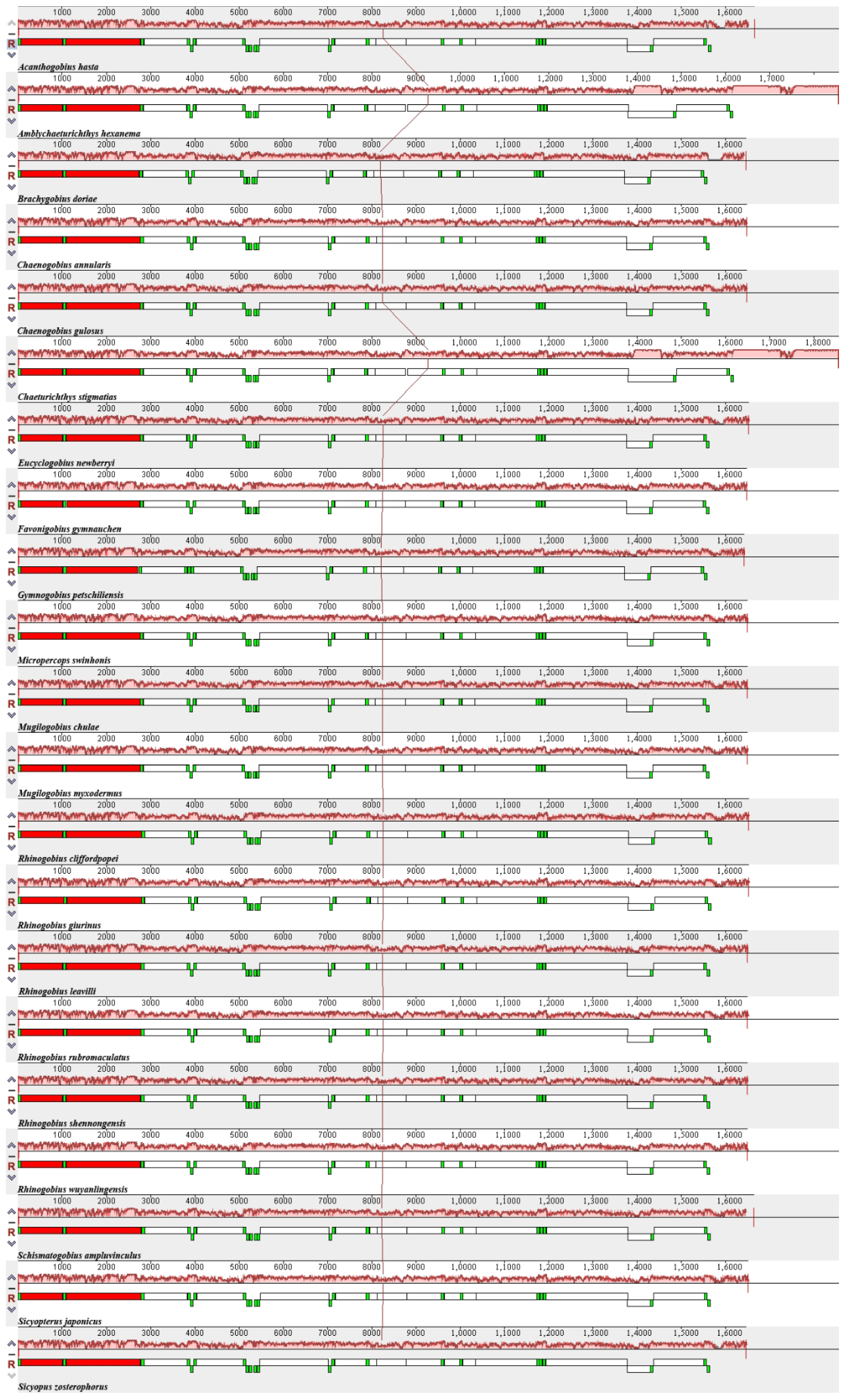
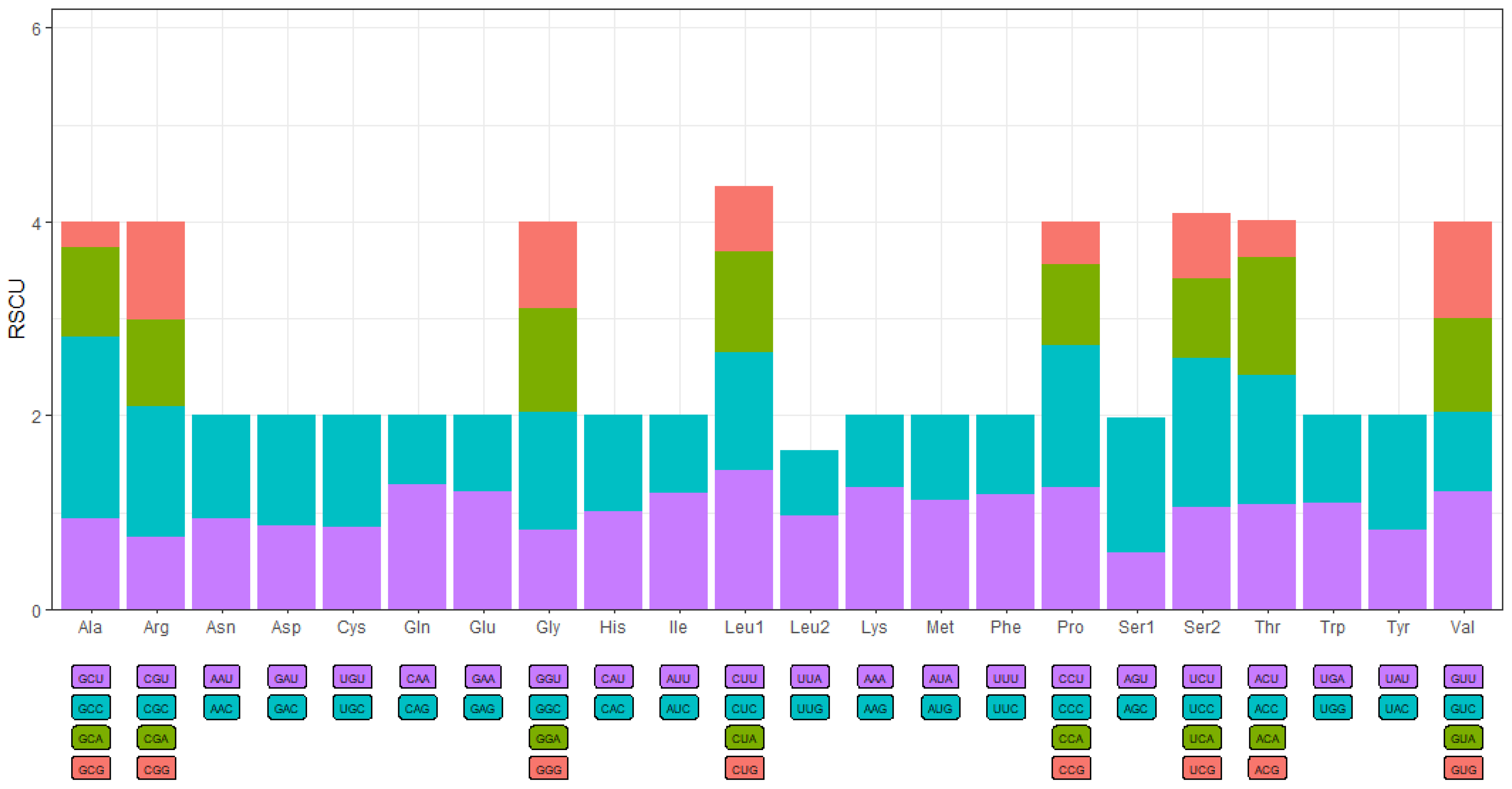
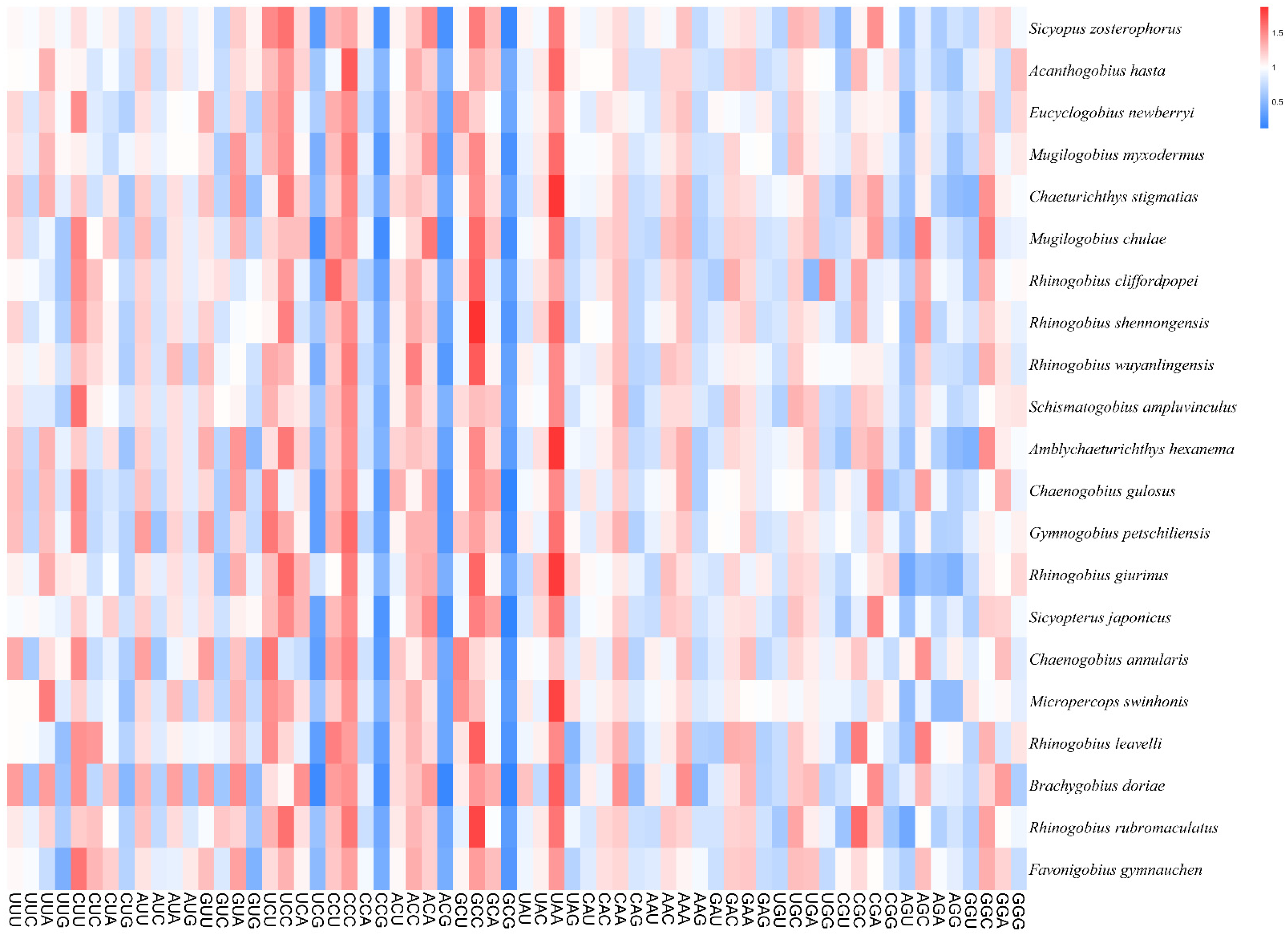
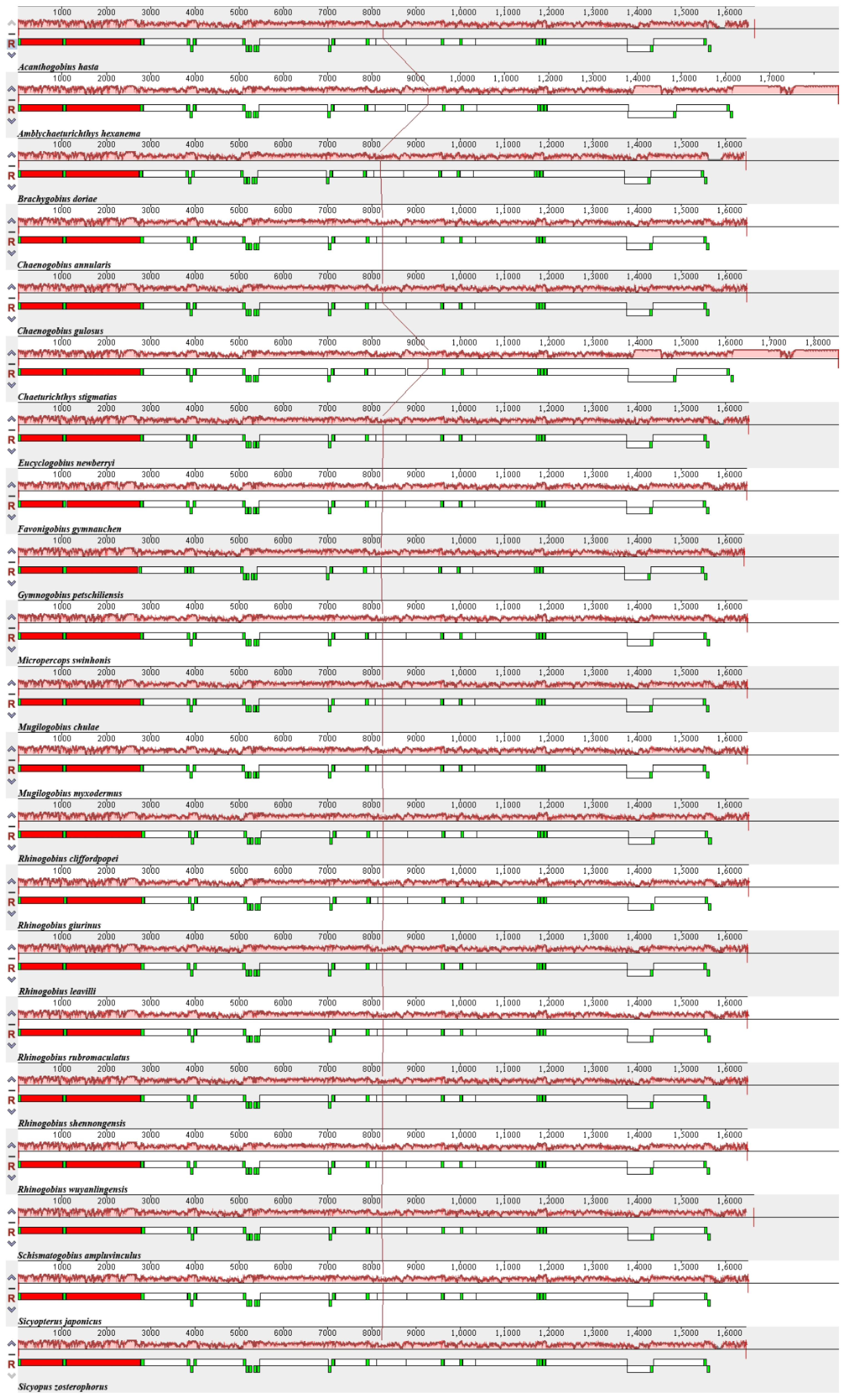
3.2. Comparison of Mitogenomes among Species
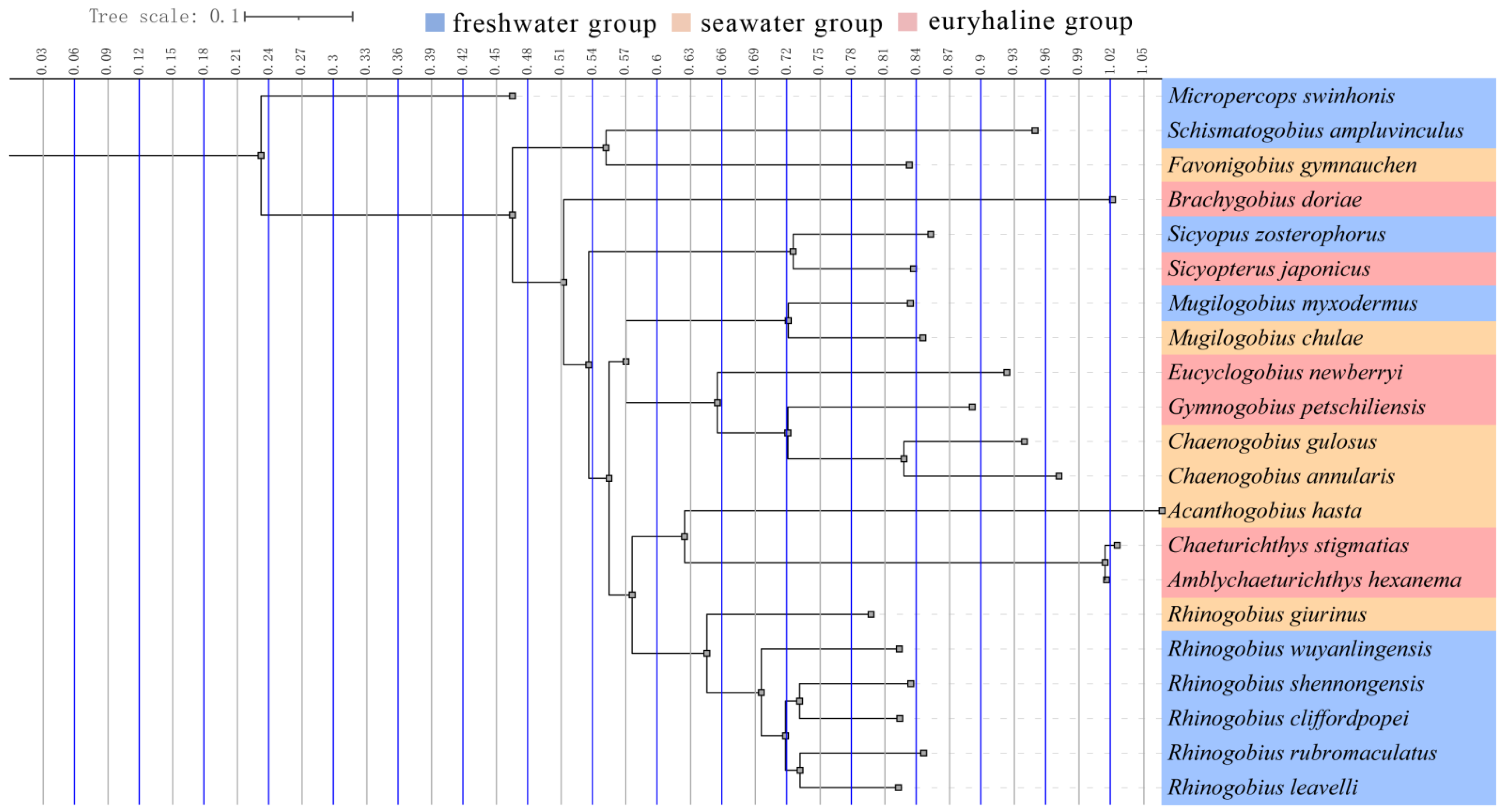
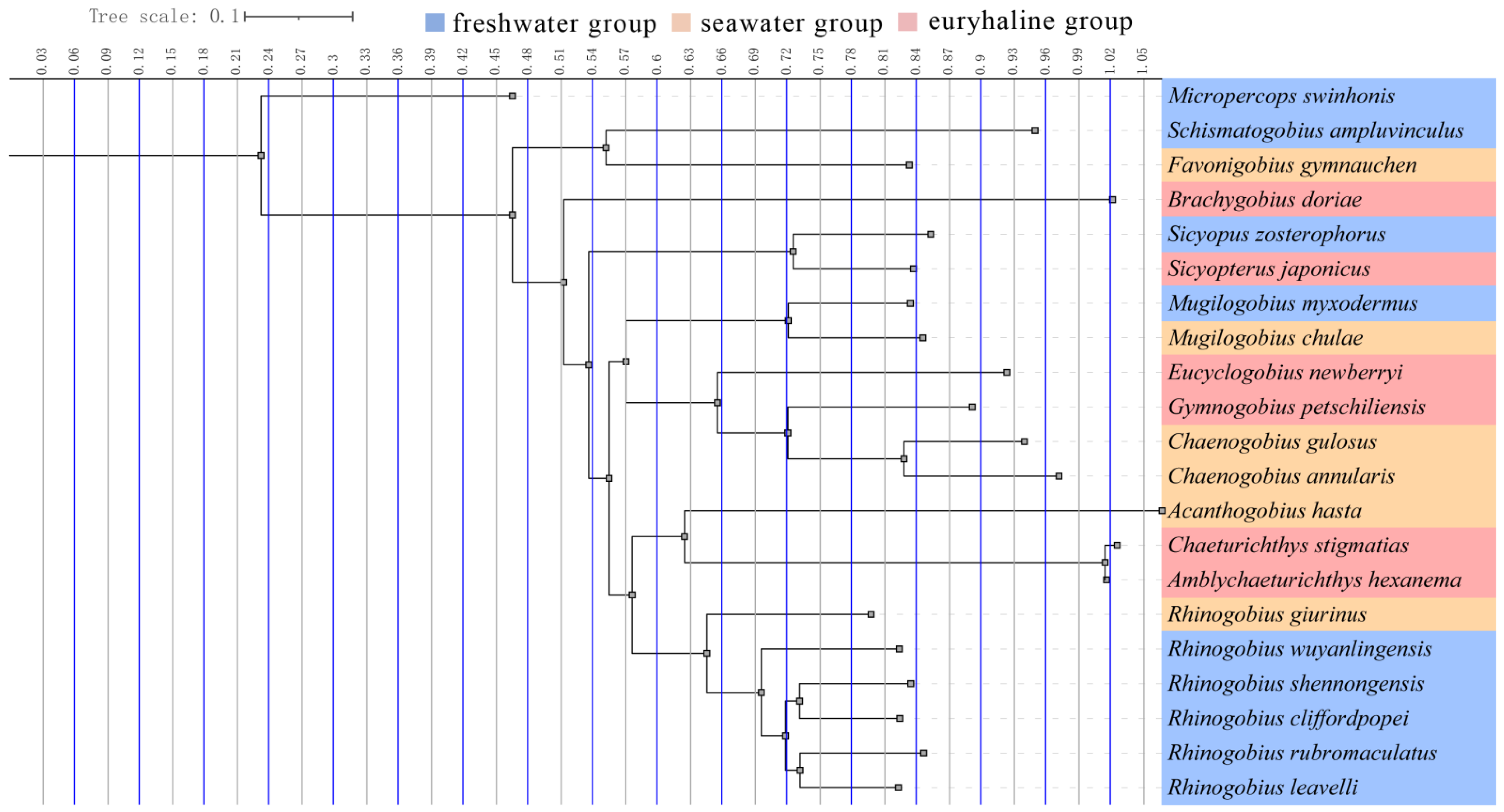
3.3. Phylogenetic Analysis
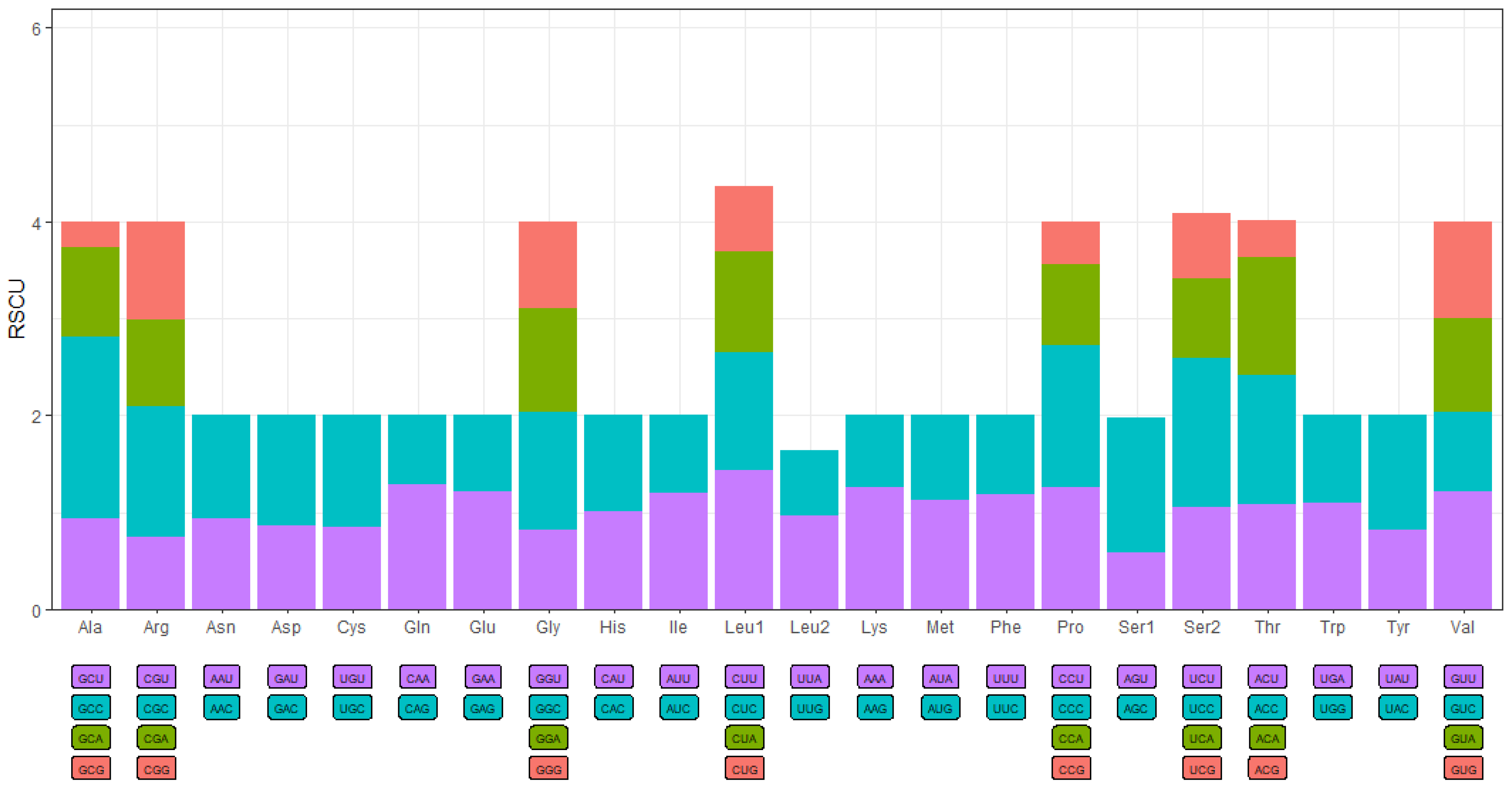
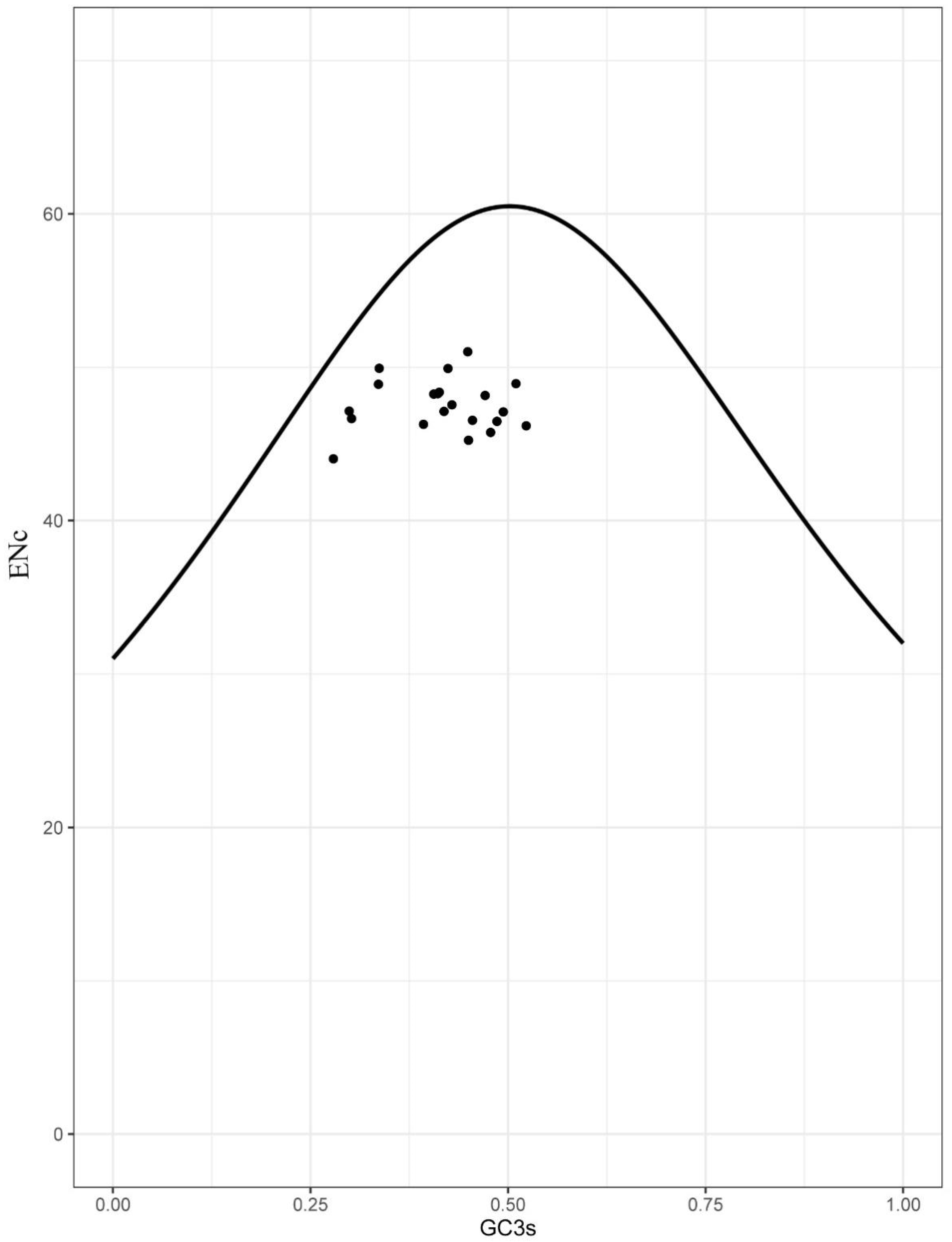
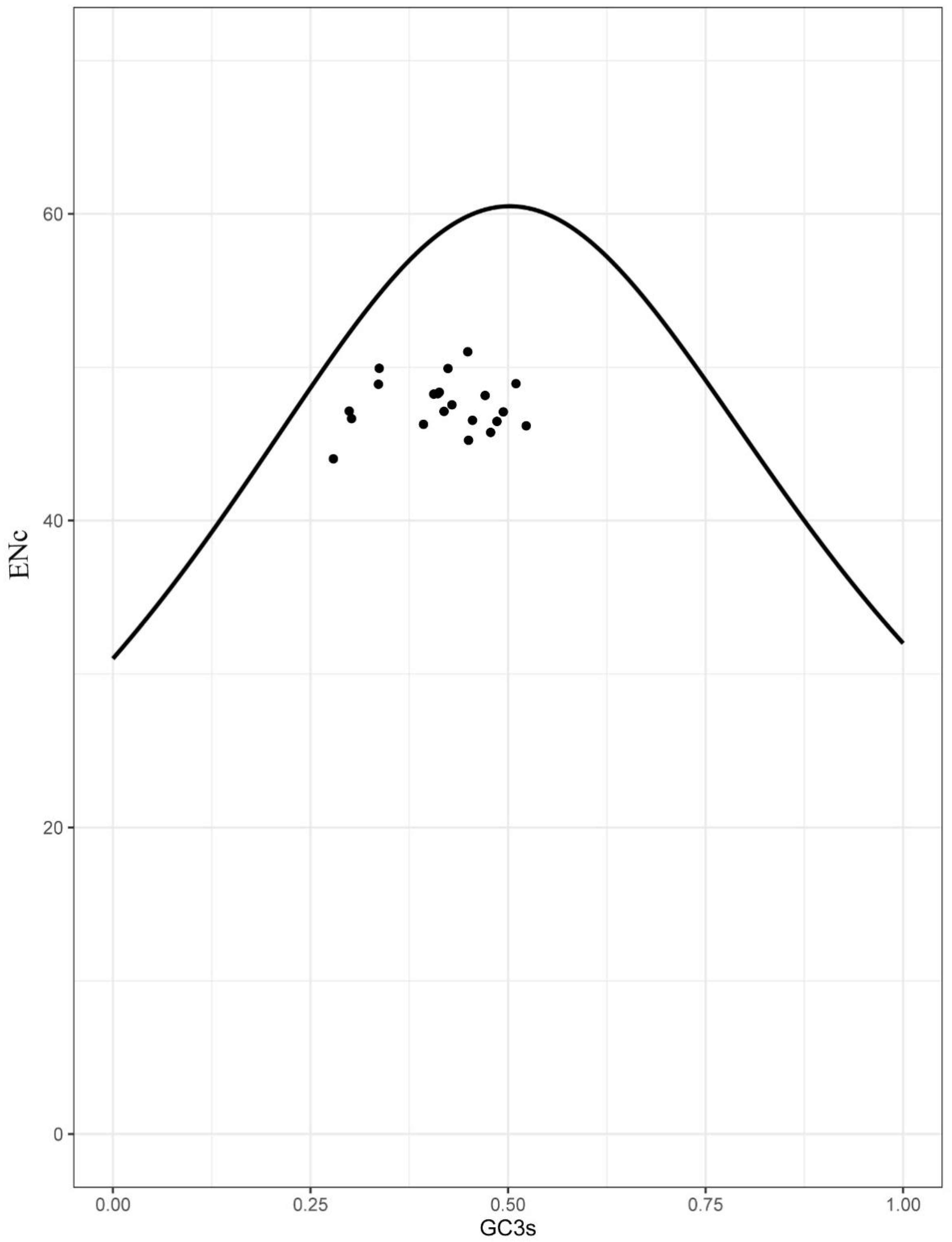
3.4. Selection Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parenti, L.R.; Nelson, J.S. Fishes of the World. Copeia 1995, 1995, 262–264. [Google Scholar] [CrossRef]
- Hubbs, C.; Moser, H.G.; Richards, W.J.; Cohen, D.M.; Fahay, M.P.; Kendall, A.W.; Richardson, S.L. Ontogeny and Systematics of Fishes. Copeia 1985, 1985, 523. [Google Scholar] [CrossRef]
- Bauchot, M.L.; Hureau, J.C. Fishes of the North-Eastern Atlantic and the Mediterranean. Unesco 1986, 61, 290. [Google Scholar]
- Van der Giezen, M.; Tovar, J.; Clark, C.G. Mitochondrion-Derived Organelles in Protists and Fungi. Int. Rev. Cytol. 2005, 244, 175–225. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell Biology of the Mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. Regulation analysis of energy metabolism. J. Exp. Biol. 1997, 200, 193–202. [Google Scholar] [CrossRef]
- Shinde, S.; Bhadra, U. A complex genome-microRNA interplay in human mitochondria. BioMed Res. Int. 2015, 2015, 206382. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, P.D.; Zhang, D.Z.; Zhang, H.B.; Tang, B.P.; Liu, Q.N.; Dai, L.S. Mitochondrial genome of the yellow catfish Pelteobagrus fulvidraco and insights into Bagridae phylogenetics. Genomics 2019, 111, 1258–1265. [Google Scholar] [CrossRef]
- Mascolo, C.; Ceruso, M.; Palma, G.; Anastasio, A.; Pepe, T.; Sordino, P. The complete mitochondrial genome of the Pink dentex Dentex gibbosus (Perciformes: Sparidae). Mitochondrial DNA Part B Resour. 2018, 3, 525–526. [Google Scholar] [CrossRef] [Green Version]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chen, Y.; Chen, Z.; He, G.; Zhong, Z.; Xue, W. The next-generation sequencing reveals the complete mitochondrial genome of Rhinogobius formosanus (Perciformes: Gobiidae). Mitochondrial DNA Part B Resour. 2020, 5, 2673–2674. [Google Scholar] [CrossRef] [PubMed]
- Dhar, D.; Dey, D.; Basu, S.; Fortunato, H. Insight into the adaptive evolution of mitochondrial genomes in intertidal chitons. J. Molluscan Stud. 2021, 87, eyab018. [Google Scholar] [CrossRef]
- Sokolova, I. Mitochondrial Adaptations to Variable Environments and Their Role in Animals’ Stress Tolerance. Integr. Comp. Biol. 2018, 58, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Lartigue, L.; Faustin, B. Mitochondria: Metabolic regulators of innate immune responses to pathogens and cell stress. Int. J. Biochem. Cell Biol. 2013, 45, 2052–2056. [Google Scholar] [CrossRef]
- Caccone, A.; Gentile, G.; Burns, C.E.; Sezzi, E.; Bergman, W.; Ruelle, M.; Saltonstall, K.; Powell, J.R. Extreme difference in rate of mitochondrial and nuclear DNA evolution in a large ectotherm, Galapagos tortoises. Mol. Phylogenetics Evol. 2004, 31, 794–798. [Google Scholar] [CrossRef]
- Harrison, R.G. Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 1989, 4, 6–11. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Saccone, C.; De Giorgi, C.; Gissi, C.; Pesole, G.; Reyes, A. Evolutionary genomics in Metazoa: The mitochondrial DNA as a model system. Gene 1999, 238, 195–209. [Google Scholar] [CrossRef]
- Lopez-Lopez, A.; Vogler, A.P. The mitogenome phylogeny of Adephaga (Coleoptera). Mol. Phylogenetics Evol. 2017, 114, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, Z.; Peng, Z. The mitochondrial genome of the water spiderArgyroneta aquatica(Araneae: Cybaeidae). Zool. Scr. 2015, 44, 179–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, Y.; Cheng, J.; Pan, D.; Lv, L.; Cumberlidge, N.; Sun, H. Phylogenetic implications of mitogenome rearrangements in East Asian potamiscine freshwater crabs (Brachyura: Potamidae). Mol. Phylogenetics Evol. 2020, 143, 106669. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, A. Comparative mitochondrial genomics within and among species of killifish. BMC Evol. Biol. 2009, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Lv, Y.; Wen, Z.; Bian, C.; Zhang, X.; Guo, S.; Shi, Q.; Li, D. The complete mitochondrial genome of the intertidal spider (Desis jiaxiangi) provides novel insights into the adaptive evolution of the mitogenome and the evolution of spiders. BMC Ecol. Evol. 2021, 21, 72. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K.; Battistuzzi, F.U. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- He, W.; Wang, N.; Tan, J.; Wang, R.; Yang, Y.; Li, G.; Guan, H.; Zheng, Y.; Shi, X.; Ye, R.; et al. Comprehensive codon usage analysis of porcine deltacoronavirus. Mol. Phylogenetics Evol. 2019, 141, 106618. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.Y.; Tsai, M.P.; Dean, J.; Lee, S.C. Molecular phylogeny of gobioid fishes (Perciformes: Gobioidei) based on mitochondrial 12S rRNA sequences. Mol. Phylogenetics Evol. 2001, 20, 390–408. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, J.M.; Van Tassell, J.L.; St Mary, C.M.; Joyeux, J.C.; Crow, K.D. Extreme gender flexibility: Using a phylogenetic framework to infer the evolution of variation in sex allocation, phylogeography, and speciation in a genus of bidirectional sex changing fishes(Lythrypnus, Gobiidae). Mol. Phylogenetics Evol. 2012, 64, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.; Freitas, L.; Nery, M.F. The role of selection in the evolution of marine turtles mitogenomes. Sci. Rep. 2020, 10, 16953. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.H.; Tian, F.; Liu, S.J.; Feng, C.G.; Zhao, K. Mitochondrial genome and phylogenetic relationship of Gymnocypris eckloni (Schizothoracinae) in Qaidam river basin. Genomics 2020, 112, 4316–4321. [Google Scholar] [CrossRef]
- Yi, J.; Wu, H.; Liu, J.; Li, J.; Lu, Y.; Zhang, Y.; Cheng, Y.; Guo, Y.; Li, D.; An, Y. Novel gene rearrangement in the mitochondrial genome of Anastatus fulloi (Hymenoptera Chalcidoidea) and phylogenetic implications for Chalcidoidea. Sci. Rep. 2022, 12, 1351. [Google Scholar] [CrossRef]
- Sun, Y.; Huang, H.; Liu, Y.; Liu, S.; Xia, J.; Zhang, K.; Geng, J. Organization and phylogenetic relationships of the mitochondrial genomes of Speiredonia retorta and other lepidopteran insects. Sci. Rep. 2021, 11, 2957. [Google Scholar] [CrossRef]
- Kim, I.C.; Kweon, H.S.; Kim, Y.J.; Kim, C.B.; Gye, M.C.; Lee, W.O.; Lee, Y.S.; Lee, J.S. The complete mitochondrial genome of the javeline goby Acanthogobius hasta (Perciformes, Gobiidae) and phylogenetic considerations. Gene 2004, 336, 147–153. [Google Scholar] [CrossRef]
- Liu, T.; Jin, X.; Wang, R.; Xu, T. Complete sequence of the mitochondrial genome of Odontamblyopus rubicundus (Perciformes: Gobiidae): Genome characterization and phylogenetic analysis. J. Genet. 2013, 92, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Mukhopadhyay, S.; Sarkar, I.; Sen, A. Comparative investigation of the various determinants that influence the codon and amino acid usage patterns in the genus Bifidobacterium. World J. Microbiol. Biotechnol. 2015, 31, 959–981. [Google Scholar] [CrossRef] [PubMed]
- Smith, R. Enhanced effective codon numbers to understand codon usage bias. BioRxiv 2019, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Sharma, S.K.; Sarkar, I.; Ray, S.D.; Pramod, P.; Kochiganti, V.H.S.; Quadros, G.; Rathore, S.S.; Singh, V.; Singh, R.P. Complete mitogenome of endemic plum-headed parakeet Psittacula cyanocephala-characterization and phylogenetic analysis. PLoS ONE 2021, 16, e0241098. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Feng, C.; Zhao, K.; Song, Z.; Zhang, Y.; Yang, L.; He, S. Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. Sci. Rep. 2016, 6, 29690. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R. Molecular signatures of natural selection. Annu. Rev. Genet. 2005, 39, 197–218. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Zhang, L.; Sun, K.; Csorba, G.; Hughes, A.C.; Jin, L.; Xiao, Y.; Feng, J. Complete mitochondrial genomes reveal robust phylogenetic signals and evidence of positive selection in horseshoe bats. BMC Ecol. Evol. 2021, 21, 199. [Google Scholar] [CrossRef]
- Shen, Y.Y.; Shi, P.; Sun, Y.B.; Zhang, Y.P. Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Res. 2009, 19, 1760–1765. [Google Scholar] [CrossRef] [Green Version]
- Muse, S.V. Examining rates and patterns of nucleotide substitution in plants. Plant Mol. Biol. 2000, 42, 25–43. [Google Scholar] [CrossRef]
- Yang, H.; Li, T.; Dang, K.; Bu, W. Compositional and mutational rate heterogeneity in mitochondrial genomes and its effect on the phylogenetic inferences of Cimicomorpha (Hemiptera: Heteroptera). BMC Genom. 2018, 19, 264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhou, S.; Wu, X.; Wei, Q.; Shang, Y.; Sun, G.; Mei, X.; Dong, Y.; Sha, W.; Zhang, H. High-altitude adaptation in vertebrates as revealed by mitochondrial genome analyses. Ecol. Evol. 2021, 11, 15077–15084. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.Y.; Liang, L.; Zhu, Z.H.; Zhou, W.P.; Irwin, D.M.; Zhang, Y.P. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 107, 8666–8671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Family | Accession Number | AT% | AT Skew | GC% | GC Skew | Length (bp) | Group |
|---|---|---|---|---|---|---|---|---|
| Acanthogobius hasta | Gobiidae | NC_006131.1 | 53.9 | 0.075 | 46.1 | −0.256 | 16,663 | seawater group (SG) |
| Favonigobius gymnauchen | Gobiidae | NC_047227.1 | 50.8 | 0.092 | 49.2 | −0.269 | 16,480 | |
| Chaenogobius gulosus | Gobiidae | NC_027193.1 | 59.0 | 0.113 | 41.0 | −0.247 | 16,477 | |
| Rhinogobius giurinus | Gobiidae | NC_022692.1 | 53.3 | 0.060 | 46.7 | −0.311 | 16,520 | |
| Chaenogobius annularis | Gobiidae | OM830225 | 58.9 | 0.145 | 41.1 | −0.204 | 16,477 | |
| Mugilogobius chulae | Gobiidae | NC_026519.1 | 54.4 | 0.081 | 45.6 | −0.279 | 16,489 | |
| Amblychaeturichthys hexanema | Gobiidae | NC_029228.1 | 54.9 | 0.093 | 45.1 | −0.256 | 18,562 | euryhaline group (EG) |
| Chaeturichthys stigmatias | Gobiidae | NC_020786.1 | 55.0 | 0.096 | 45.0 | −0.254 | 18,562 | |
| Eucyclogobius newberryi | Gobiidae | NC_028288.1 | 53.0 | 0.163 | 47.0 | −0.226 | 16,523 | |
| Sicyopterus japonicus | Gobiidae | NC_045074.1 | 53.7 | 0.050 | 46.3 | −0.284 | 16,504 | |
| Gymnogobius petschiliensis | Gobiidae | NC_008743.1 | 57.4 | 0.140 | 42.6 | −0.228 | 16,424 | |
| Brachygobius doriae | Gobiidae | NC_037142.1 | 60.8 | 0.049 | 39.2 | −0.272 | 16,472 | |
| Rhinogobius cliffordpopei | Gobiidae | NC_029252.1 | 49.4 | 0.097 | 50.6 | −0.291 | 16,525 | freshwater group (FG) |
| Rhinogobius leavelli | Gobiidae | NC_044964.1 | 51.8 | 0.080 | 48.2 | −0.310 | 16,499 | |
| Rhinogobius rubromaculatus | Gobiidae | NC_037144.1 | 51.5 | 0.089 | 48.5 | −0.306 | 16,503 | |
| Mugilogobius myxodermus | Gobiidae | NC_036070.1 | 54.2 | 0.082 | 45.8 | −0.304 | 16,495 | |
| Schismatogobius ampluvinculus | Gobiidae | NC_035717.1 | 52.3 | 0.098 | 47.7 | −0.262 | 16,639 | |
| Sicyopus zosterophorus | Gobiidae | NC_058982.1 | 55.0 | 0.057 | 45.0 | −0.281 | 16,471 | |
| Micropercops swinhonis | Odontobutidae | NC_021763.1 | 57.8 | 0.124 | 42.2 | −0.225 | 16,493 | |
| Rhinogobius shennongensis | Gobiidae | OM961050 | 52.3 | 0.044 | 47.7 | −0.312 | 16,500 | |
| Rhinogobius wuyanlingensis | Gobiidae | OM961051 | 53.2 | 0.070 | 46.8 | −0.306 | 16,491 |
| Gene | Nucleotide Positions | Size (bp) | Strand | Intergenic Nucleotide | Start | Stop |
|---|---|---|---|---|---|---|
| tRNAPHE | 1–68 | 68 | + | |||
| 12s rRNA | 69–1014 | 946 | + | 0 | ||
| tRNAVAL | 1015–1086 | 72 | + | 0 | ||
| 16s rRNA | 1091–2767 | 1677 | + | 4 | ||
| tRNALEU | 2768–2842 | 75 | + | 0 | ||
| nd1 | 2844–3818 | 975 | + | 1 | ATG | TAA |
| tRNAILE | 3823–3893 | 71 | + | 4 | ||
| tRNAGLN | 3894–3964 | 71 | − | 0 | ||
| tRNAMET | 3964–4032 | 69 | + | −1 | ||
| nd2 | 4033–5079 | 1065 | + | 0 | ATG | TAA |
| tRNATRP | 5079–5150 | 72 | + | −1 | ||
| tRNAALA | 5153–5221 | 69 | − | 2 | ||
| tRNAASN | 5223–5295 | 73 | − | 1 | ||
| tRNACYS | 5331–5395 | 65 | − | 35 | ||
| tRNATYR | 5396–5466 | 71 | − | 0 | ||
| co1 | 5468–7021 | 1554 | + | 1 | GTG | TAA |
| tRNASER | 7022–7092 | 71 | − | 0 | ||
| tRNAASP | 7096–7167 | 72 | + | 3 | ||
| co2 | 7171–7861 | 691 | + | 3 | ATG | T |
| tRNALYS | 7862–7936 | 75 | + | 0 | ||
| atp8 | 7938–8102 | 165 | + | 1 | ATG | TAG |
| atp6 | 8096–8779 | 684 | + | −7 | ATG | TAA |
| co3 | 8779–9563 | 785 | + | −1 | ATG | TA |
| tRNAGLY | 9563–9631 | 69 | + | −1 | ||
| nd3 | 9632–9980 | 349 | + | 0 | ATG | T |
| tRNAARG | 9981–10049 | 69 | + | 0 | ||
| nd4l | 10050–10346 | 297 | + | 0 | ATG | TAA |
| nd4 | 10340–11720 | 1381 | + | −7 | ATG | T |
| tRNAHIS | 11721–11789 | 69 | + | 0 | ||
| tRNASER | 11790–11856 | 67 | + | 0 | ||
| tRNALEU | 11860–11932 | 73 | + | 3 | ||
| nd5 | 11933–13768 | 1836 | + | 0 | ATG | TAG |
| nd6 | 13765–14286 | 522 | − | −4 | ATG | TAA |
| tRNAGLU | 14287–14355 | 69 | − | 0 | ||
| cyb | 14361–15498 | 1138 | + | 5 | ATG | TAG |
| tRNATHR | 15502–15573 | 72 | + | 3 | ||
| tRNAPRO | 15574–15643 | 70 | − | 0 |
| Gene | Nucleotide Positions | Size (bp) | Strand | Intergenic Nucleotide | Start | Stop |
|---|---|---|---|---|---|---|
| tRNAPHE | 1–68 | 68 | + | |||
| 12s rRNA | 69–1017 | 949 | + | 0 | ||
| tRNAVAL | 1018–1089 | 72 | + | 0 | ||
| 16s rRNA | 1098–2778 | 1681 | + | 8 | ||
| tRNALEU | 2779–2853 | 75 | + | 0 | ||
| nd1 | 2854–3828 | 975 | + | 0 | ATG | TAA |
| tRNAILE | 3831–3900 | 70 | + | 2 | ||
| tRNAGLN | 3900–3970 | 71 | − | −1 | ||
| tRNAMET | 3970–4038 | 69 | + | −1 | ||
| nd2 | 4039–5085 | 1047 | + | 0 | ATG | TAA |
| tRNATRP | 5087–5157 | 71 | + | 1 | ||
| tRNAALA | 5160–5228 | 69 | − | 2 | ||
| tRNAASN | 5230–5302 | 73 | − | 1 | ||
| tRNACYS | 5335–5399 | 65 | − | 32 | ||
| tRNATYR | 5400–5469 | 70 | − | 0 | ||
| co1 | 5471–7024 | 1554 | + | 1 | GTG | TAA |
| tRNASER | 7025–7095 | 71 | − | 0 | ||
| tRNAASP | 7099–7170 | 72 | + | 3 | ||
| co2 | 7174–7864 | 691 | + | 3 | ATG | T |
| tRNALYS | 7865–7940 | 76 | + | 0 | ||
| atp8 | 7942–8106 | 165 | + | 1 | ATG | TAA |
| atp6 | 8100–8783 | 684 | + | −7 | ATG | TAA |
| co3 | 8783–9567 | 785 | + | −1 | ATG | TA |
| tRNAGLY | 9567–9638 | 72 | + | −1 | ||
| nd3 | 9639–9987 | 349 | + | 0 | ATG | T |
| tRNAARG | 9988–10,056 | 69 | + | 0 | ||
| nd4l | 10,057–10,353 | 297 | + | 0 | ATG | TAA |
| nd4 | 10,347–11,727 | 1381 | + | −7 | ATG | T |
| tRNAHIS | 11,728–11,796 | 69 | + | 0 | ||
| tRNASER | 11,797–11,864 | 68 | + | 0 | ||
| tRNALEU | 11,869–11,941 | 73 | + | 4 | ||
| nd5 | 11,942–13,780 | 1839 | + | −4 | ATG | TAA |
| nd6 | 13,777–14,298 | 522 | − | −4 | ATG | TAG |
| tRNAGLU | 14,299–14,367 | 69 | − | 0 | ||
| cyb | 14,373–15,512 | 1140 | + | 5 | ATG | T |
| tRNATHR | 15,514–15,585 | 72 | + | 1 | ||
| tRNAPRO | 15,585–15,645 | 61 | − | −1 |
| Gene | Nucleotide Positions | Size (bp) | Strand | Intergenic Nucleotide | Start | Stop |
|---|---|---|---|---|---|---|
| tRNAPHE | 1–68 | 68 | + | |||
| 12s rRNA | 69–1019 | 951 | + | 0 | ||
| tRNAVAL | 1019–1090 | 72 | + | −1 | ||
| 16s rRNA | 1099–2774 | 1676 | + | 8 | ||
| tRNALEU | 2775–2848 | 74 | + | 0 | ||
| nd1 | 2849–3823 | 975 | + | 0 | ATG | TAA |
| tRNAILE | 3828–3897 | 70 | + | 4 | ||
| tRNAGLN | 3897–3967 | 71 | − | −1 | ||
| tRNAMET | 3967–4035 | 69 | + | −1 | ||
| nd2 | 4036–5082 | 1047 | + | 0 | ATG | TAA |
| tRNATRP | 5084–5154 | 71 | + | 1 | ||
| tRNAALA | 5157–5225 | 69 | − | 2 | ||
| tRNAASN | 5227–5299 | 73 | − | 1 | ||
| tRNACYS | 5332–5397 | 66 | − | 2 | ||
| tRNATYR | 5398–5467 | 70 | − | 0 | ||
| co1 | 5469–7022 | 1554 | + | 1 | GTG | TAA |
| tRNASER | 7023–7093 | 71 | − | 0 | ||
| tRNAASP | 7097–7168 | 72 | + | 3 | ||
| co2 | 7172–7862 | 691 | + | 3 | ATG | T |
| tRNALYS | 7863–7938 | 76 | + | 0 | ||
| atp8 | 7940–8104 | 165 | + | 1 | ATG | TAA |
| atp6 | 8098–8781 | 684 | + | −7 | ATG | TAA |
| co3 | 8781–9565 | 785 | + | −1 | ATG | TA |
| tRNAGLY | 9565–9635 | 71 | + | −1 | ||
| nd3 | 9636–9984 | 349 | + | 0 | ATG | T |
| tRNAARG | 9985–10,053 | 69 | + | 0 | ||
| nd4l | 10,054–10,350 | 297 | + | 0 | ATG | TAA |
| nd4 | 10,344–11,724 | 1381 | + | −7 | ATG | T |
| tRNAHIS | 11,725–11,792 | 68 | + | 0 | ||
| tRNASER | 11,793–11,860 | 68 | + | 0 | ||
| tRNALEU | 11,865–11,937 | 73 | + | 4 | ||
| nd5 | 11,938–13,776 | 1839 | + | 0 | ATG | TAG |
| nd6 | 13,773–14,294 | 522 | − | −4 | ATG | TAG |
| tRNAGLU | 14,295–14,363 | 69 | − | 0 | ||
| cyb | 14,369–15,508 | 1140 | + | 5 | ATG | T |
| tRNATHR | 15,510–15,581 | 72 | + | 1 | ||
| tRNAPRO | 15,581–15,650 | 139 | − | −1 |
| Gene | Model Compared | |2ΔlnL| | p-Value | ωSG | ωEG | ωFG |
|---|---|---|---|---|---|---|
| atp6 | M2a_rel vs. CmC | 10.16444 | 0.017218814 * | 0.13766 | 0.06356 | 0.08155 |
| atp8 | 28.09846 | 3.46325×10-6 ** | 0.30112 | 0.36634 | 0.11016 | |
| co1 | 123.17652 | 1.59679×10-26 ** | 0.10628 | 0.13944 | 0.15688 | |
| co2 | 15.34283 | 0.001545921 ** | 0.07167 | 0.04816 | 0.06300 | |
| co3 | 3.31304 | 0.34583 | 0.09589 | 0.09700 | 0.10508 | |
| cyb | 234.47697 | 1.48869×10-50 ** | 0.06100 | 0.10580 | 0.06614 | |
| nd1 | 8.34667 | 0.039364856 * | 0.10092 | 0.04505 | 0.13131 | |
| nd2 | 3.35000 | 0.000227486 ** | 0.10087 | 0.07880 | 0.11073 | |
| nd3 | 5.49261 | 0.13908 | 0.13504 | 0.07199 | 0.14217 | |
| nd4 | 3.93641 | 0.26841 | 0.12748 | 0.10171 | 0.13955 | |
| nd4l | 8.604689 | 0.035035806 * | 0.08711 | 0.06757 | 0.08709 | |
| nd5 | 271.95414 | 1.16596×10-58 ** | 0.12279 | 0.11736 | 0.15599 | |
| nd6 | 1407.00957 | 8.8747×10-305 ** | 0.07906 | 0.22650 | 0.15444 |
| Gene | Model | 2ΔLNL | p-Value | Positively Selected Sites (BEB Analysis) | Foreground Branch | Background Branch |
|---|---|---|---|---|---|---|
| nd6 | Model A Null Model | 0 | 1 | 2 S 0.963 *; 100 G 0.977 *; 139 F 0.989 * | EG | FG and SG |
| atp8 | 0 | 1 | 7 S 0.999 **; 25 V 0.988 *; 36 E 0.978 *; 41 N 1.000 **; 51 S 0.966 * | EG | FG and SG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, Y.; Wang, X.; Liu, G.; Wu, X.; Wei, Q.; Sun, G.; Mei, X.; Dong, Y.; Sha, W.; Zhang, H. Adaptability and Evolution of Gobiidae: A Genetic Exploration. Animals 2022, 12, 1741. https://doi.org/10.3390/ani12141741
Shang Y, Wang X, Liu G, Wu X, Wei Q, Sun G, Mei X, Dong Y, Sha W, Zhang H. Adaptability and Evolution of Gobiidae: A Genetic Exploration. Animals. 2022; 12(14):1741. https://doi.org/10.3390/ani12141741
Chicago/Turabian StyleShang, Yongquan, Xibao Wang, Gang Liu, Xiaoyang Wu, Qinguo Wei, Guolei Sun, Xuesong Mei, Yuehuan Dong, Weilai Sha, and Honghai Zhang. 2022. "Adaptability and Evolution of Gobiidae: A Genetic Exploration" Animals 12, no. 14: 1741. https://doi.org/10.3390/ani12141741
APA StyleShang, Y., Wang, X., Liu, G., Wu, X., Wei, Q., Sun, G., Mei, X., Dong, Y., Sha, W., & Zhang, H. (2022). Adaptability and Evolution of Gobiidae: A Genetic Exploration. Animals, 12(14), 1741. https://doi.org/10.3390/ani12141741