
The Mitochondrial Genome of Cylicocyclus elongatus (Strongylida: Strongylidae) and Its Comparative Analysis with Other Cylicocyclus Species

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Parasites and Molecular Identification of Specimens
2.2. The Amplification and Annotation of C. elongatus Complete Mt Genome
2.3. Comparative Analyses of Cylicocyclus Species and P. imparidentatum Mt Genomes
2.4. Phylogenetic Analyses of 23 Strongylidae Nematodes
3. Results
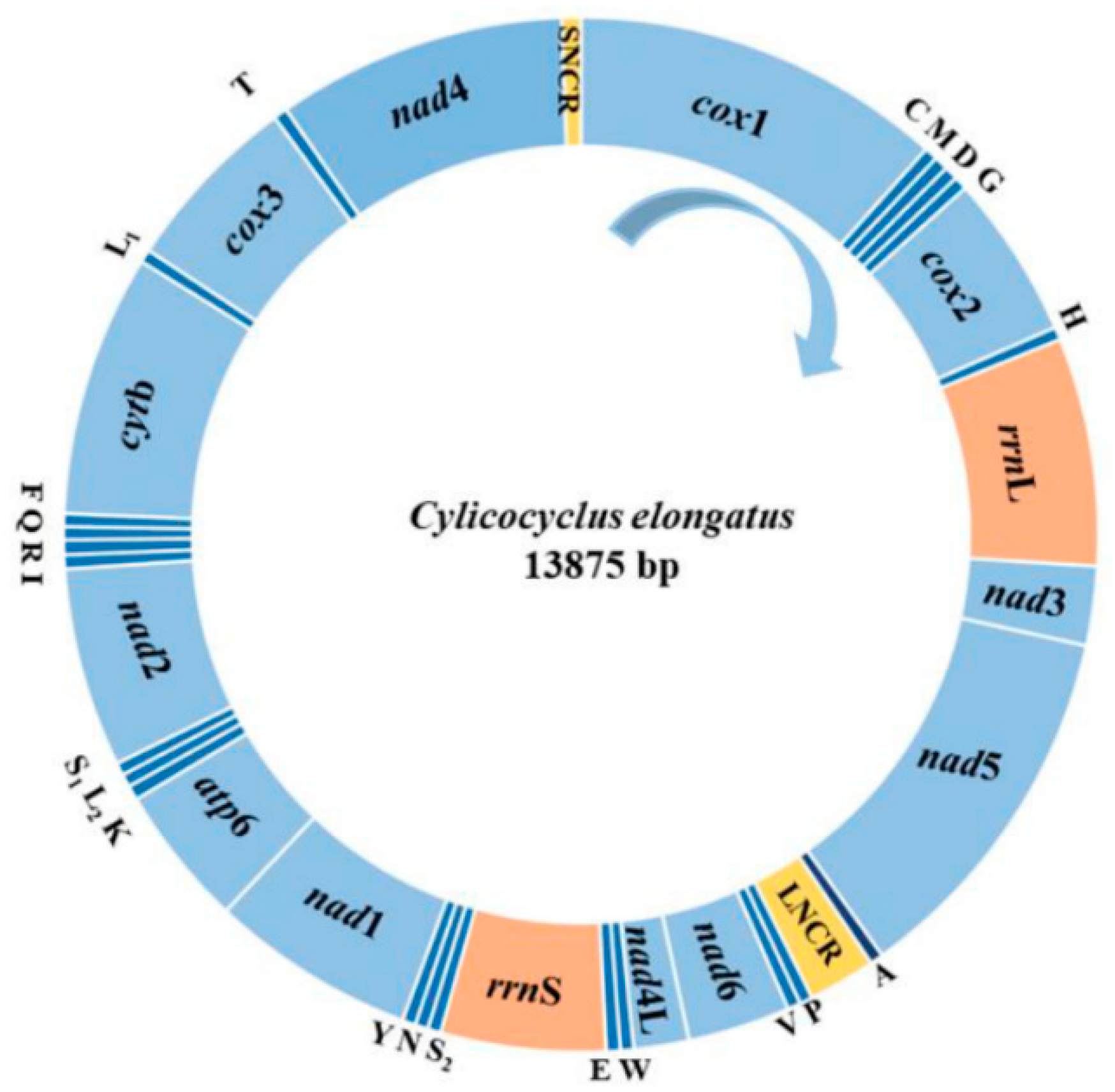
3.1. The Annotation of C. elongatus Complete Mt Genome
3.2. Comparative Analyses of Mt Genomes among Cylicocyclus Species and P. imparidentatum
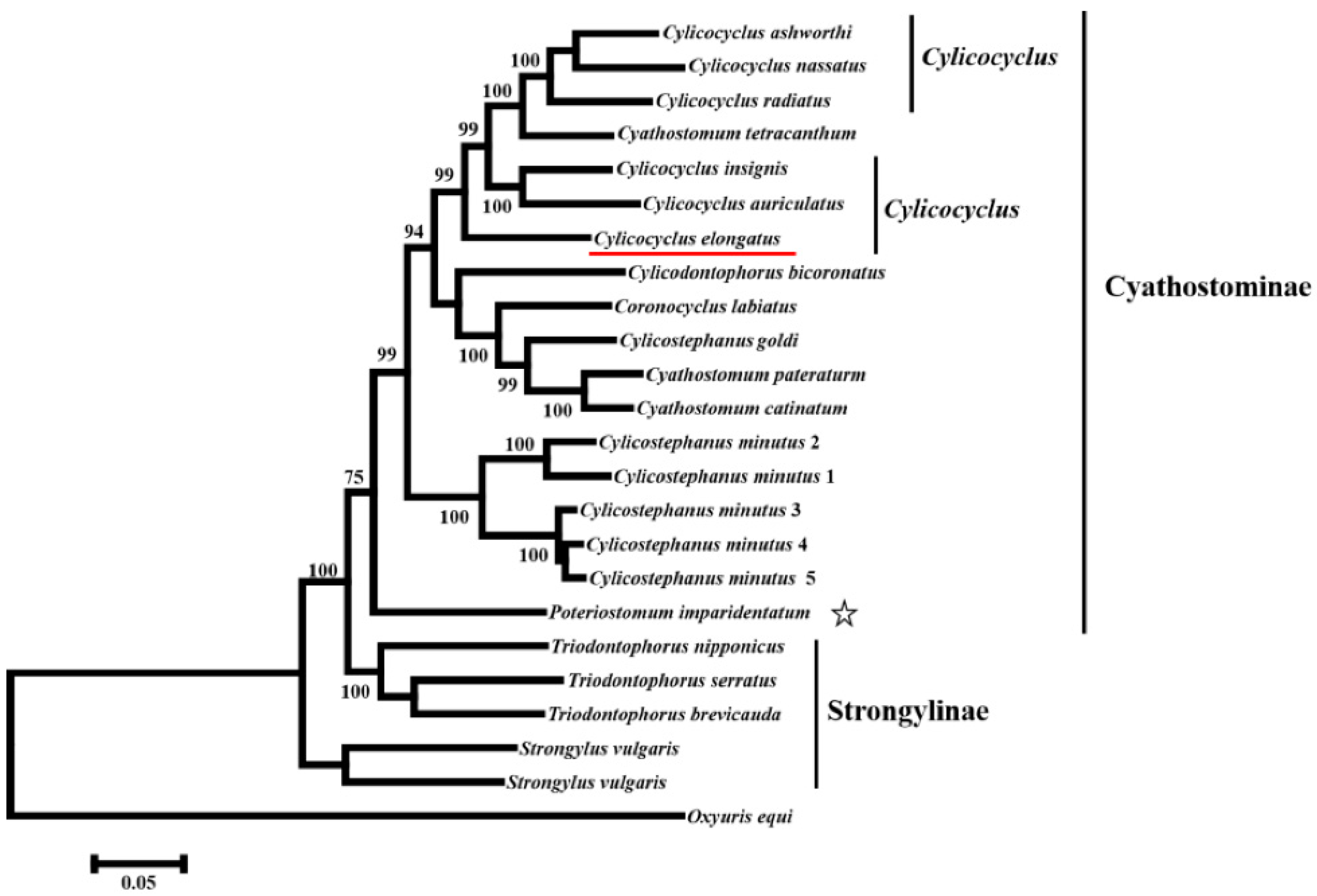
3.3. Phylogenetic Analyses of 23 Species in the Family Strongylidae
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lichtenfels, J.R.; Kharchenko, V.A.; Dvojnos, G.M. Illustrated identification keys to strongylid parasites (Strongylidae:Nematoda) of horses, zebras and asses (Equidae). Vet. Parasitol. 2008, 156, 4–161. [Google Scholar] [CrossRef] [PubMed]
- Corning, S. Equine cyathostomins: A review of biology, clinical significance and therapy. Parasites Vectors 2009, 2, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jota Baptista, C.; Sós, E.; Madeira de Carvalho, L. Gastrointestinal parasitism in przewalski horses (Equus ferus przewalskii). Acta Parasitol. 2021, 66, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Bredtmann, C.M.; Krücken, J.; Murugaiyan, J.; Balard, A.; Hofer, H.; Kuzmina, T.A.; von Samson-Himmelstjerna, G.V. Concurrent proteomic fingerprinting and molecular analysis of cyathostomins. Proteomics 2019, 19, e1800290. [Google Scholar] [CrossRef] [PubMed]
- Morariu, S.; Mederle, N.; Badea, C.; Dărăbuş, G.; Ferrari, N.; Genchi, C. The prevalence, abundance and distribution of cyathostomins (small stongyles) in horses from western Romania. Vet. Parasitol. 2016, 223, 205–209. [Google Scholar] [CrossRef]
- Hartwich, G. Zum Strongylus tetracanthus-problem und zursystematik der Cyathostominea (Nematoda:Strongyloidea). Mitt. Zool. Mus. Berl. 1986, 62, 61–102. [Google Scholar] [CrossRef]
- Zhang, L.P.; Kong, F.Y. Review of the systematics of Cyathostominea (Nematoda:Strongylidae). Acta Zootaxon. Sin. 2002, 27, 435–446. (In Chinese) [Google Scholar]
- Bu, Y.; Niu, H.; Zhang, L. Phylogenetic analysis of the genus Cylicocyclus (Nematoda: Strongylidae) based on nuclear ribosomal sequence data. Acta Parasitol. 2013, 58, 167–173. [Google Scholar] [CrossRef]
- Hu, M.; Gasser, R.B. Mitochondrial genomes of parasitic nematodes–progress and perspectives. Trends Parasitol. 2006, 22, 78–84. [Google Scholar] [CrossRef]
- Jex, A.R.; Hall, R.S.; Littlewood, D.T.; Gasser, R.B. An integrated pipeline for next generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010, 38, 522–533. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, Y.; Yang, X.; Qiu, J.H.; Duan, H.; Xu, W.W.; Chang, Q.C.; Wang, C.R. Mitochondrial DNA evidence supports the hypothesis that Triodontophorus species belong to Cyathostominae. Front. Microbiol. 2017, 8, 1444. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Wang, X.X.; Ma, X.X.; Zhang, Z.H.; Lan, Z.; Qiu, Y.Y.; Wang, S.; Song, M.X.; Wang, C.R. Characterization of the complete mitochondrial genomes of Coronocyclus labiatus and Cylicodontophorus bicoronatus: Comparison with Strongylidae species and phylogenetic implication. Vet. Parasitol. 2021, 290, 109359. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Wu, C.Y.; Song, H.Q.; Wei, S.J.; Xu, M.J.; Lin, R.Q.; Zhao, G.H.; Huang, S.Y.; Zhu, X.Q. Comparative analyses of the complete mitochondrial genomes of Ascaris lumbricoides and Ascaris suum from humans and pigs. Gene 2012, 492, 110–116. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, M.; Sun, Y.; Bu, Y. Characterization and phylogenetic analysis of the first complete mitochondrial genome of Cylicocyclus radiatus. Vet. Parasitol. 2020, 281, 109097. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; Bott, N.J.; Chilton, N.B.; Hunt, P.; Beveridge, I. Toward practical, DNA-based diagnostic methods for parasitic nematodes of livestock-bionomic and biotechnological implications. Biotechnol. Adv. 2008, 26, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Qiu, J.H.; Zhang, B.B.; Su, X.; Fu, X.; Yue, D.M.; Wang, C.R. Complete mitochondrial genome of parasitic nematode Cylicocyclus nassatus and comparative analyses with Cylicocyclus insigne. Exp. Parasitol. 2017, 172, 18–22. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Higgins, D.G. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Gao, Y.; Wang, X.X.; Li, Y.; Gao, J.F.; Wang, C.R. The complete mitochondrial genome of Cylicocylus ashworthi (Rhabditida: Cyathostominae). Mitochondrial DNA Part B 2019, 4, 1225–1226. [Google Scholar] [CrossRef] [Green Version]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Zeng, M.; Diao, P.; Wang, X.; Li, Q.; Li, Y.; Gao, Y.; Wang, C. Comparative analyses of the complete mitochondrial genomes of Cyathostomum pateratum and Cyathostomum catinatum provide new molecular data for the evolution of Cyathostominae nematodes. J. Helminthol. 2019, 93, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Zhao, L.; Song, H.Q.; Zhao, G.H.; Cai, J.Z.; Zhao, Q.; Zhu, X.Q. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasites Vectors 2014, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.F.; Liu, G.H.; Duan, H.; Gao, Y.; Zhang, Y.; Chang, Q.C.; Fang, M.; Wang, C.R. Complete mitochondrial genomes of Triodontophorus serratus and Triodontophorus nipponicus, and their comparison with Triodontophorus brevicauda. Exp. Parasitol. 2017, 181, 88–93. [Google Scholar] [CrossRef]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomics of parasitic nematodes of socio-economic importance: Recent progress, and implications for population genetics and systematics. Adv. Parasitol. 2004, 56, 133–212. [Google Scholar]



{kind=link}
{kind=link}
| Genes/Regions | Positions and Sequence Lengths (bp) | Initiation and Stop Codons | Intergenic Nucleotides |
|---|---|---|---|
| cox1 | 1–1578 (1578) | ATT/TAA | 0 |
| tRNA-Cys (C) | 1579–1634 (56) | – | 0 |
| tRNA-Met (M) | 1648–1706 (59) | – | 13 |
| tRNA-Asp (D) | 1707–1765 (58) | – | 0 |
| tRNA-Gly (G) | 1787–1843 (57) | – | 22 |
| cox2 | 1844–2539 (696) | ATT/TAA | 0 |
| tRNA-His (H) | 2540–2593 (54) | – | 0 |
| rrnL | 2601–3568 (968) | – | 7 |
| nad3 | 3572–3907 (336) | ATT/TAA | 4 |
| nad5 | 3918–5501 (1584) | ATT/TAG | 10 |
| tRNA-Ala (A) | 5501–5556 (56) | – | −1 |
| LNCR | 5557–5842 (286) | – | 0 |
| tRNA-Pro (P) | 5843–5897 (55) | – | 0 |
| tRNA-Val (V) | 5931–5985 (55) | – | 33 |
| nad6 | 5986–6420 (435) | ATT/TAA | 0 |
| nad4L | 6473–6706 (234) | ATT/TAA | 52 |
| tRNA-Trp (W) | 6732–6787 (56) | – | 21 |
| tRNA-Glu (E) | 6815–6874 (60) | – | 27 |
| rrnS | 6877–7575 (699) | – | 2 |
| tRNA-SerUCN (S2) | 7576–7631 (56) | – | 0 |
| tRNA-Asn (N) | 7630–7684 (55) | – | −2 |
| tRNA-Tyr (Y) | 7693–7748 (56) | – | 8 |
| nad1 | 7749–8621 (873) | TTG/TAA | 0 |
| atp6 | 8632–9231 (600) | ATT/TAA | 10 |
| tRNA-Lys (K) | 9248–9309 (62) | – | 16 |
| tRNA-LeuUUR (L2) | 9328–9382 (55) | – | 18 |
| tRNA-SerAGN (S1) | 9383–9435 (53) | – | 0 |
| nad2 | 9436–10,281 (846) | TTG/TAA | 0 |
| tRNA-Ile (I) | 10,288–10,346 (59) | – | 6 |
| tRNA-Arg (R) | 10,374–10,436 (63) | – | 27 |
| tRNA-Gln (Q) | 10,444–10,498 (55) | – | 7 |
| tRNA-Phe (F) | 10,505–10,559 (55) | – | 6 |
| cytb | 10,560–11,672 (1113) | ATT/TAA | 0 |
| tRNA-LeuCUN (L1) | 11,683–11,737 (55) | – | 11 |
| cox3 | 11,738–12,506 (769) | ATT/T | 0 |
| tRNA-Thr (T) | 12,507–12,564 (58) | – | 0 |
| nad4 | 12,565–13,794 (1230) | TTG/TAA | 0 |
| SNCR | 13,795–13,875 (81) | – | 0 |
| Total size (bp) | 13,875 | – |
| Species | Identity Nts (%) | ||||||
|---|---|---|---|---|---|---|---|
| C. el | C. as | C. i | C. au | C. n | C. r | P. i | |
| Total Size (bp) | 13,828 | 13,817 | 13,876 | 13,836 | 13,831 | 13,846 | 13,875 |
| C. as | 87.1 | – | – | – | – | – | – |
| C. i | 88.1 | 88.2 | – | – | – | – | – |
| C. au | 87.2 | 87.4 | 89.6 | – | – | – | – |
| C. n | 87.1 | 90.7 | 88.0 | 87.2 | – | – | – |
| C. r | 87.4 | 90.7 | 88.0 | 87.6 | 89.4 | – | – |
| P. i | 84.3 | 82.9 | 83.6 | 82.9 | 83.0 | 83.3 | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Zhang, Z.; Wang, C.; Zhao, K. The Mitochondrial Genome of Cylicocyclus elongatus (Strongylida: Strongylidae) and Its Comparative Analysis with Other Cylicocyclus Species. Animals 2022, 12, 1571. https://doi.org/10.3390/ani12121571
Gao Y, Zhang Z, Wang C, Zhao K. The Mitochondrial Genome of Cylicocyclus elongatus (Strongylida: Strongylidae) and Its Comparative Analysis with Other Cylicocyclus Species. Animals. 2022; 12(12):1571. https://doi.org/10.3390/ani12121571
Chicago/Turabian StyleGao, Yuan, Zhonghuai Zhang, Chunren Wang, and Kai Zhao. 2022. "The Mitochondrial Genome of Cylicocyclus elongatus (Strongylida: Strongylidae) and Its Comparative Analysis with Other Cylicocyclus Species" Animals 12, no. 12: 1571. https://doi.org/10.3390/ani12121571
APA StyleGao, Y., Zhang, Z., Wang, C., & Zhao, K. (2022). The Mitochondrial Genome of Cylicocyclus elongatus (Strongylida: Strongylidae) and Its Comparative Analysis with Other Cylicocyclus Species. Animals, 12(12), 1571. https://doi.org/10.3390/ani12121571