
Metabolomic Profiles of Multidrug-Resistant Salmonella Typhimurium from Humans, Bovine, and Porcine Hosts

 and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolate Collection, Screening, Identification, and Growth
2.2. Integron and AMR Testing
2.3. Isolate Growth and Extraction for Proteomic and Metabolomics Profiling
2.4. Metabolomic Profiling by UPLC- and GC–MS
2.5. Statistical Analysis (MetaboAnalyst 4.0)
3. Results
3.1. AMR Patterns and Integrons
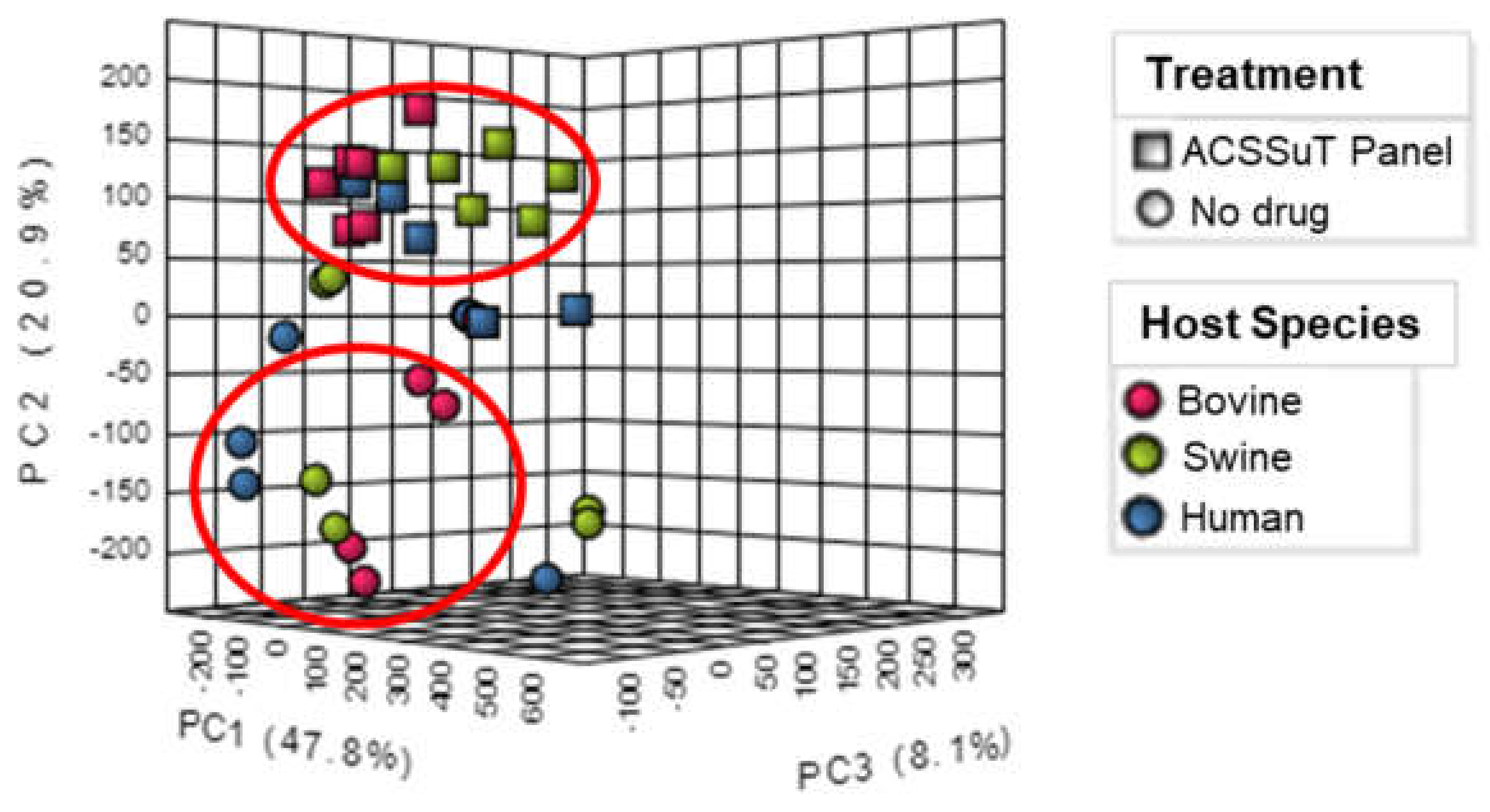
3.2. Metabolite Expression by Drug Treatment and Host Species
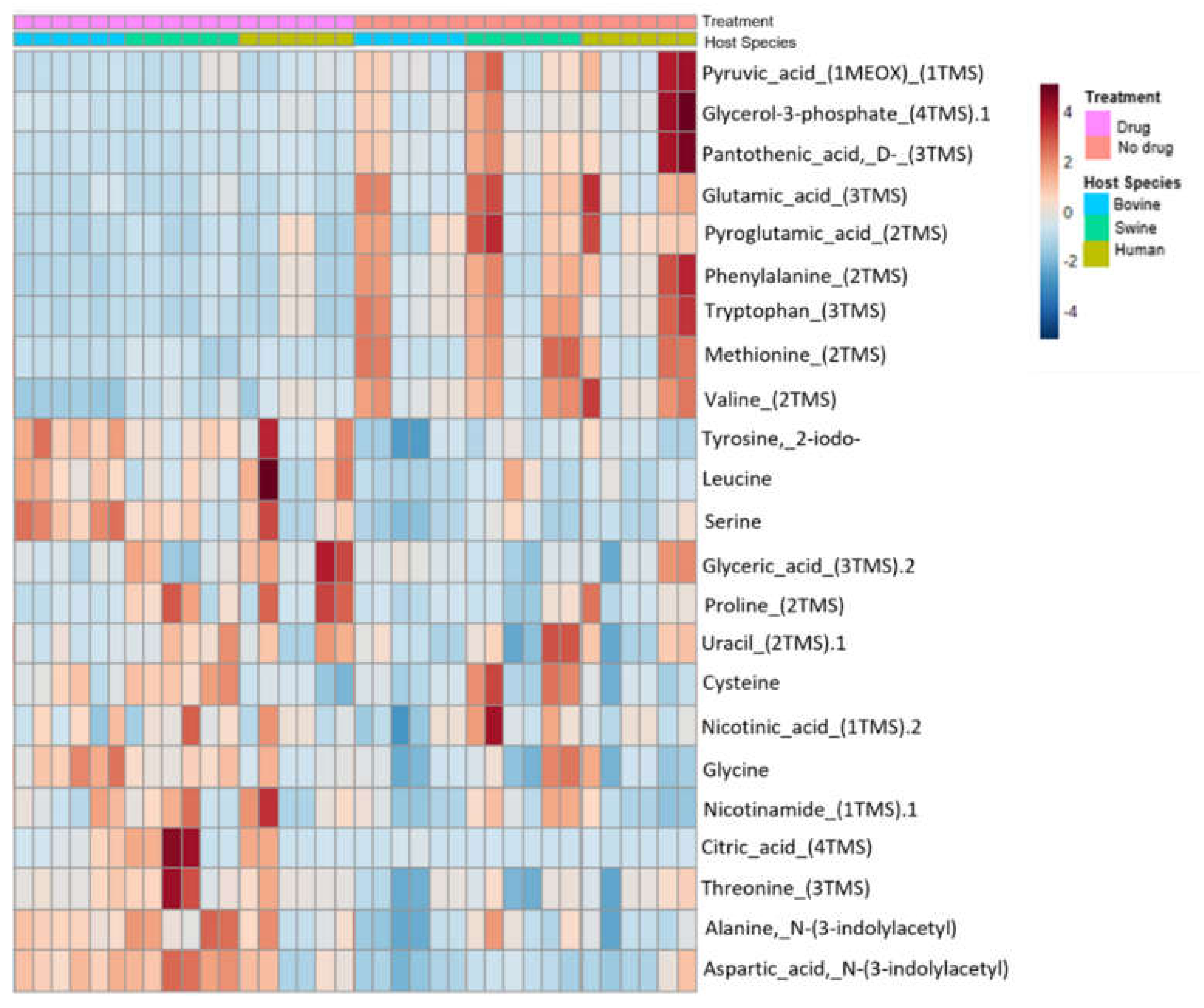
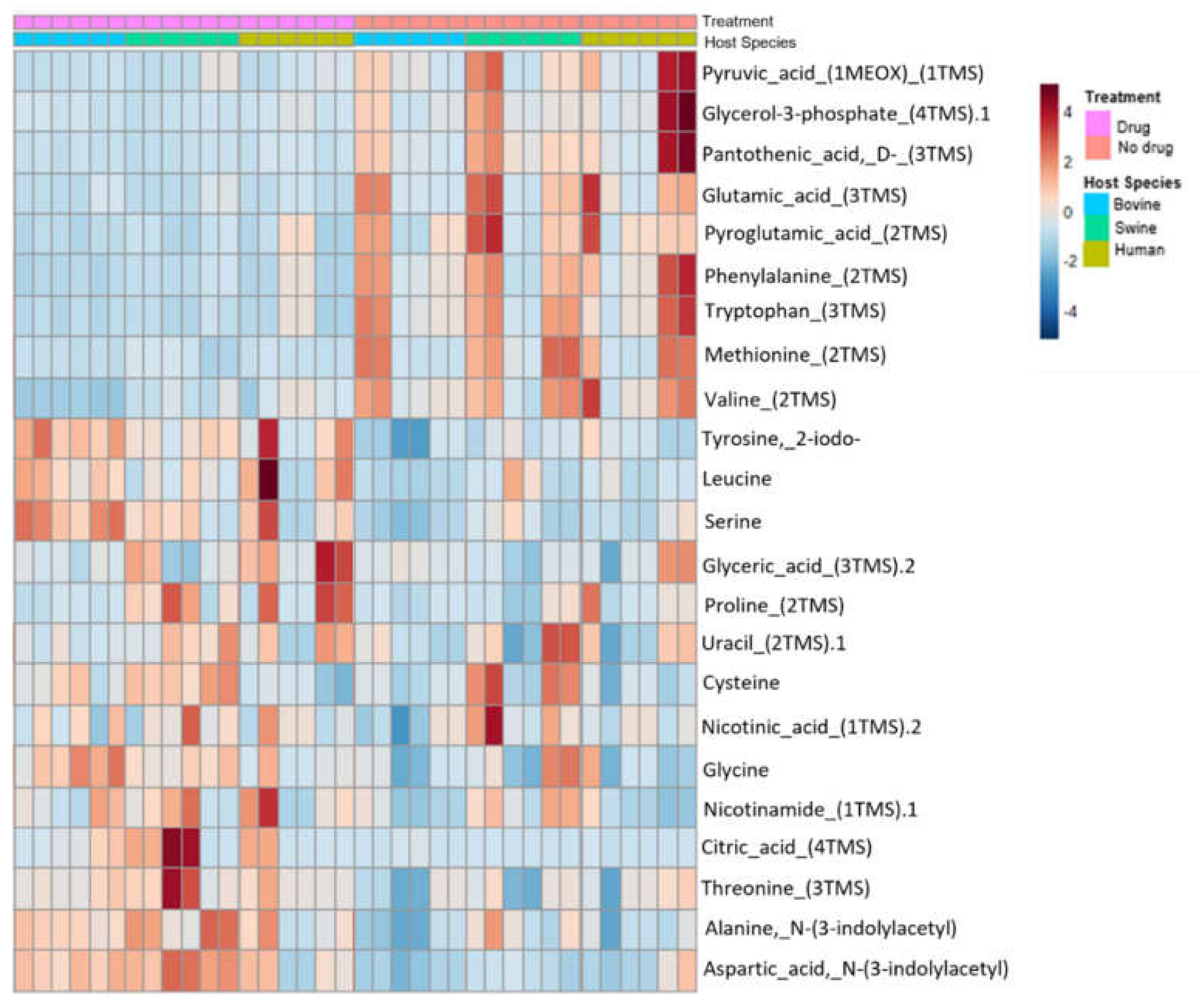
3.3. Metabolite Expression and Matched Metabolic Pathways
3.4. Univariate Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC. What Exactly Is Antibiotic Resistance? Centers for Disease Control and Prevention. 2020. Available online: https://www.cdc.gov/drugresistance/about.html (accessed on 21 January 2022).
- CDC. National Action Plan for Combating Antibiotic-Resistant Bacteria. Centers for Disease Control and Prevention. 2015. Available online: https://www.cdc.gov/drugresistance/pdf/national_action_plan_for_combating_antibotic-resistant_bacteria.pdf (accessed on 21 January 2022).
- Van Belkum, A.; Burnham, C.D.; Rossen, J.W.A.; Mallard, F.; Rochas, O.; Dunne, W.M. Innovative and rapid antimicrobial susceptibility testing systems. Nat. Rev. Microbiol. 2020, 18, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Hoerr, V.; Duggan, G.; Zbytnuik, L.; Poon, K.H.; Grobe, C.; Neugebauer, U.; Methling, K.; Loffler, B.; Vogel, H. Characterization and prediction of the mechanism of action of antibiotics through NMR metabolomics. BMC Microbiol. 2016, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C. Where will new antibiotics come from? Nat. Rev. Microbiol. 2003, 1, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Dailey, A.; Saha, J.; Zaidi, F.; Abdirahman, H.; Haymond, A.; Alem, F.; Hakami, R.; Couch, R. VOC fingerprints: Metabolomic signatures of biothreat agents with and without antibiotic resistance. Sci. Rep. 2020, 10, 11746. [Google Scholar] [CrossRef]
- Agbaje, M.; Begum, R.H.; Oyekunle, M.A.; Ojo, O.E.; Adenubi, O.T. Evolution of Salmonella nomenclature: A critical note. Folia Microbiol. 2011, 56, 497–503. [Google Scholar] [CrossRef]
- European Food Safety Authority & European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2017/2018. EFSA J. 2020, 18, 6007. [Google Scholar]
- Rao, S.; Maddox, C.; Hoien-Dalen, P.; Lanka, S.; Weigel, R. Diagnostic accuracy of class 1 integron PCR method in detection of antibiotic resistance in Salmonella isolates from swine production systems. J. Clin. Microbiol. 2008, 46, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Broeckling, C.; Afsar, F.; Neumann, S.; Ben-Hur, A.; Prenni, J. RAMClust: A Novel Feature Clustering Method Enables Spectral-Matching-Based Annotation for Metabolomics Data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef]
- Plumb, R.S.; Johnson, K.A.; Rainville, P.; Smith, B.W.; Wilson, I.D.; Castro-Perez, J.M.; Nicholson, J.K. UPLC/MSE; A new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun. Mass Spectrom. 2006, 20, 1989–1994. [Google Scholar] [CrossRef]
- Smith, C.; Want, E.; Elizabeth, J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear PSeak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J., Jr. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Shlaes, D.M.; Sahm, D.; Opiela, C.; Spellberg, B. Commentary: The FDA reboot of antibiotic development. Antimicrob. Agents Chemother. 2013, 57, 4605–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aries, M.L.; Cloninger, M.J. NMR Hydrophilic Metabolomic Analysis of Bacterial Resistance Pathways Using Multivalent Antimicrobials with Challenged and Unchallenged Wild Type and Mutated Gram-Positive Bacteria. Int. J. Mol. Sci. 2021, 22, 13606. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B. The future of antibiotics. Crit. Care 2014, 18, 228. [Google Scholar] [CrossRef] [Green Version]
- Lindon, J.C.; Holmes, E.; Nicholson, J.K. Metabonomics Techniques and Applications to Pharmaceutical Research & Development. Pharm. Res. 2006, 23, 1076–1088. [Google Scholar] [CrossRef]
- Dörries, K.; Schlueter, R.; Lalk, M. Impact of antibiotics with various target sites on the metabolome of Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 7151–7163. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Li, W.; Sun, L.; Lin, Z.; Jiang, Y.; Ling, Y.; Lin, X. Comparative metabolomics shows the metabolic profiles fluctuate in multi-drug resistant Escherichia coli strains. J. Proteom. 2019, 207, 103468. [Google Scholar] [CrossRef]
- Franklin, T.J.; Snow, G.A. Biochemical Mechanisms of Resistance to Antimicrobial Drugs. In Biochemistry and Molecular Biology of Antimicrobial Drug Action, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 149–174. [Google Scholar]
- Su, Y.-B.; Peng, B.; Li, H.; Cheng, Z.-X.; Zhang, T.-T.; Zhu, J.-X.; Li, D.; Li, M.-Y.; Ye, J.-Z.; Du, C.-C.; et al. Pyruvate Cycle Increases Aminoglycoside Efficacy and Provides Respiratory Energy in Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Grube, C.; Hervé, R. A Quantitative Spectrophotometric Assay to Monitor the TRNA-Dependent Pathway for Lipid Aminoacylation In Vitro. J. Biomol. Screen. 2016, 21, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, L.; Shears, R.; Fingleton, C.; Alvarez, L.; Waters, E.; Clarke, J.; Bricio-Moreno, L.; Campbell, C.; Yadav, A.K.; Razvi, F.; et al. Impaired Alanine Transport or Exposure to D-Cycloserine Increases the Susceptibility of MRSA to β-Lactam Antibiotics. J. Infect. Dis. 2020, 221, 1000–1016. [Google Scholar] [CrossRef]
- Alreshidi, M.; Dunstan, R.; Macdonald, M.; Smith, N.; Gottfries, J.; Roberts, T. Metabolomic and Proteomic Responses of Staphylococcus aureus to Prolonged Cold Stress. J. Proteom. 2015, 121, 44–55. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Oliveria, K.; de Lima, L.A.; Cobacho, N.B.; Dias, S.C.; Franco, O.L. Mechanisms of Antibacterial Resistance: Shedding Some Light on These Obscure Processes? In Antibiotic Resistance: Mechanisms and New Antimicrobial Approaches; Kon, K., Rai, M., Eds.; Academic Press: Cambridge, MA, USA, 2016; Chapter 2; pp. 19–35. [Google Scholar]

{kind=link}
{kind=link}
| Drug Panel | MIC |
|---|---|
| Ampicillin | 32 µg/mL |
| Chloramphenicol | 32 µg/mL |
| Streptomycin | 64 µg/mL |
| Sulfisoxazole | 512 µg/mL |
| Tetracycline | 16 µg/mL |
| All Hosts | Bovine | Swine | Human | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Metabolic Pathway | Impact | p-Value | FDR | Metabolite | Fold Change (FD/ND) | p-Value | FDR | Fold Change (FD/ND) | p-Value | FDR | Fold Change (FD/ND) | p-Value | FDR | Fold Change (FD/ND) | p-Value | FDR |
| Aminoacyl-tRNA biosynthesis | 0.2 | 1.93 × 10−7 | 1.66 × 10−5 | Phenylalanine | 0.565 | 0.0000581 | 0.00043499 | 0.433 | 0.0021645 | 0.0096991 | 0.737 | 0.535 | ||||
| Cysteine | 1.515 | 0.0096309 | 0.023444 | 0.118 | 0.0021645 | 0.0096991 | 0.228 | 0.0021645 | 0.022605 | 1.400 | ||||||
| Glycine | 1.858 | 0.00000394 | 0.0000655 | 2.003 | 0.0021645 | 0.0096991 | 1.812 | 1.723 | ||||||||
| Aspartate | 3.077 | 3.06 × 10−8 | 0.00000283 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 2.178 | ||||||
| Serine | 2.837 | 8.22 × 10−8 | 0.00000426 | 4.469 | 0.0021645 | 0.0096991 | 1.755 | 0.0021645 | 0.022605 | 2.240 | ||||||
| Methionine | 0.594 | 0.020464 | 0.042605 | 0.498 | 0.0021645 | 0.0096991 | 0.588 | 0.708 | ||||||||
| Valine | 0.675 | 0.0064022 | 0.017165 | 0.391 | 0.0021645 | 0.0096991 | 0.925 | 0.782 | ||||||||
| Alanine | 2.065 | 3.06 × 10−8 | 0.00000283 | 2.149 | 0.0021645 | 0.0096991 | 2.240 | 0.0021645 | 0.022605 | 1.910 | ||||||
| Leucine | 0.087 | 0.0023492 | 0.0080271 | 0.106 | 0.015152 | 0.041482 | 0.063 | 0.0021645 | 0.022605 | 0.116 | ||||||
| Threonine | 2.233 | 2.2 × 10−10 | 0.000000285 | 2.065 | 0.0021645 | 0.0096991 | 3.156 | 0.0021645 | 0.022605 | 1.834 | ||||||
| Tryptophan | 2.143 | 0.0000164 | 0.00017127 | 1.789 | 0.0021645 | 0.0096991 | 2.671 | 1.647 | ||||||||
| Tyrosine | 2.183 | 1.48 × 10−8 | 0.00000212 | 2.932 | 0.0021645 | 0.0096991 | 2.054 | 0.0021645 | 0.022605 | 1.959 | ||||||
| Proline | 2.150 | 0.00075777 | 0.0033152 | 1.153 | 3.432 | 2.041 | ||||||||||
| Glutamate | 0.321 | 0.0000239 | 0.00021944 | 0.353 | 0.497 | 0.243 | ||||||||||
| Pantothenate and CoA biosynthesis | 0.144 | 0.00304 | 0.10686 | Pantothenate | 0.414 | 0.000000 | 0.000005 | 0.440 | 0.0021645 | 0.0096991 | 0.389 | 0.0021645 | 0.022605 | 0.429 | ||
| Valine | 0.675 | 0.006402 | 0.017165 | 0.391 | 0.0021645 | 0.0096991 | 0.925 | 0.782 | ||||||||
| Aspartate | 3.077 | 0.000000 | 0.000003 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 1.300 | ||||||
| Cysteine | 1.515 | 0.009631 | 0.023444 | 0.118 | 0.0021645 | 0.0096991 | 0.228 | 0.0021645 | 0.022605 | 1.400 | ||||||
| Pyruvate | 0.458 | 0.002642 | 0.008841 | 0.382 | 0.008658 | 0.026381 | 1.570 | 0.0021645 | 0.022605 | 0.394 | ||||||
| Uracil | 1.477 | 0.020464 | 0.042605 | 0.977 | 1.848 | 1.474 | ||||||||||
| Glycine, serine, and threonine metabolism | 0.456 | 0.00373 | 0.10686 | Serine | 2.837 | 0.000000 | 0.000004 | 4.469 | 0.0021645 | 0.0096991 | 1.755 | 0.0021645 | 0.022605 | 2.240 | ||
| Glycine | 1.858 | 0.000004 | 0.000066 | 2.003 | 0.0021645 | 0.0096991 | 1.812 | 1.723 | ||||||||
| Aspartate | 3.077 | 0.000000 | 0.000003 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 1.300 | ||||||
| Glycerate | 1.691 | 0.000503 | 0.002423 | 0.631 | 0.008658 | 0.026381 | 2.225 | 2.473 | ||||||||
| Threonine | 2.233 | 0.000000 | 0.000000 | 2.065 | 0.0021645 | 0.0096991 | 3.156 | 0.0021645 | 0.022605 | 1.834 | ||||||
| Pyruvate | 0.458 | 0.002642 | 0.008841 | 0.382 | 0.008658 | 0.026381 | 1.570 | 0.0021645 | 0.022605 | 0.394 | ||||||
| Tryptophan | 2.143 | 0.000016 | 0.000171 | 1.789 | 0.0021645 | 0.0096991 | 2.671 | 1.647 | ||||||||
| Glutathione metabolism | 0.118 | 0.01061 | 0.2144 | Glycine | 1.858 | 0.000004 | 0.000066 | 2.003 | 0.0021645 | 0.0096991 | 1.812 | 1.723 | ||||
| Cysteine | 1.515 | 0.009631 | 0.023444 | 0.118 | 0.0021645 | 0.0096991 | 0.228 | 0.0021645 | 0.022605 | 1.400 | ||||||
| Pyroglutamic Acid | 0.679 | 0.005177 | 0.014605 | 0.613 | 0.748 | 0.693 | ||||||||||
| Glutamate | 0.321 | 0.000024 | 0.000219 | 0.353 | 0.497 | 0.243 | ||||||||||
| Nicotinate and Nicotinamide metabolism | 0.066 | 0.0125 | 0.2144 | Aspartate | 3.077 | 0.000000 | 0.000003 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 1.300 | ||
| Glycerone phosphate | 0.582 | 0.000001 | 0.000027 | 0.644 | 0.004329 | 0.016886 | 0.579 | 0.0021645 | 0.022605 | 0.536 | ||||||
| Nicotinamide | 1.858 | 0.000245 | 0.001366 | 1.505 | 1.666 | 2.495 | ||||||||||
| Nicotinate | 1.435 | 0.000007 | 0.000098 | 1.587 | 0.008658 | 0.026381 | 1.455 | 1.832 | ||||||||
| Glyoxylate and dicarboxylate metabolism | 0.055 | 0.0269 | 0.38631 | Citrate | 7.934 | 0.000024 | 0.000219 | 2.799 | 20.017 | 6.503 | ||||||
| Glycerate | 1.691 | 0.000503 | 0.002423 | 0.631 | 0.008658 | 0.026381 | 2.225 | 2.473 | ||||||||
| Glycine | 1.858 | 0.000004 | 0.000066 | 2.003 | 0.0021645 | 0.0096991 | 1.812 | 1.723 | ||||||||
| Glutamate | 0.321 | 0.000024 | 0.000219 | 0.353 | 0.497 | 0.243 | ||||||||||
| Serine | 2.837 | 0.000000 | 0.000004 | 4.469 | 0.0021645 | 0.0096991 | 1.755 | 0.0021645 | 0.022605 | 2.240 | ||||||
| Pyruvate | 0.458 | 0.002642 | 0.008841 | 0.382 | 0.008658 | 0.026381 | 1.570 | 0.0021645 | 0.022605 | 0.394 | ||||||
| beta-Alanine Metabolism | 0 | 0.0458 | 0.43434 | Aspartate | 3.077 | 0.000000 | 0.000003 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 1.300 | ||
| Pantothenate | 0.414 | 0.000000 | 0.000005 | 0.440 | 0.0021645 | 0.0096991 | 0.389 | 0.0021645 | 0.022605 | 0.429 | ||||||
| Uracil | 1.477 | 0.020464 | 0.042605 | 0.977 | 1.848 | 1.474 | ||||||||||
| Valine, leucine, and isoleucine biosynthesis | 0.107 | 0.0475 | 0.43434 | Threonine | 2.233 | 0.000000 | 0.000000 | 2.065 | 0.0021645 | 0.0096991 | 3.156 | 0.0021645 | 0.022605 | 1.834 | ||
| Leucine | 0.087 | 0.002349 | 0.008027 | 0.106 | 0.015152 | 0.041482 | 0.063 | 0.0021645 | 0.022605 | 0.116 | ||||||
| Pyruvate | 0.458 | 0.002642 | 0.008841 | 0.382 | 0.008658 | 0.026381 | 1.570 | 0.0021645 | 0.022605 | 0.394 | ||||||
| Valine | 0.675 | 0.006402 | 0.017165 | 0.391 | 0.0021645 | 0.0096991 | 0.925 | 0.782 | ||||||||
| Alanine, aspartate and glutamate metabolism | 0.45 | 0.0475 | 0.43434 | Aspartate | 3.077 | 0.000000 | 0.000003 | 3.174 | 0.0021645 | 0.0096991 | 3.883 | 0.0021645 | 0.022605 | 1.300 | ||
| Alanine | 2.065 | 0.000000 | 0.000003 | 2.149 | 0.0021645 | 0.0096991 | 2.240 | 0.0021645 | 0.022605 | 1.910 | ||||||
| Glutamate | 0.321 | 0.000024 | 0.000219 | 0.353 | 0.497 | 0.243 | ||||||||||
| Pyruvate | 0.458 | 0.002642 | 0.008841 | 0.382 | 0.008658 | 0.026381 | 1.570 | 0.0021645 | 0.022605 | 0.394 | ||||||
 Non-significant metabolites.
Non-significant metabolites.| Metabolite | p-Value | FDR |
|---|---|---|
| 2-Piperidinecarboxylic_acid_1MEOX_2TMS | 0.00958 | 0.04464 |
| Adenine_1TMS | 0.00275 | 0.02104 |
| Alanine, N-3-indolylacetyl | 0.00002 | 0.00135 |
| Aspartic acid, N-3-indolylacetyl | 0.00000 | 0.00032 |
| Butanoic acid,_3-hydroxy-0.2 | 0.00064 | 0.00847 |
| Butanoic_acid, 4-hydroxy-_2TMS | 0.00035 | 0.00576 |
| Cinnamic_acid, 2-hydroxy-, trans- | 0.000003 | 0.00040 |
| Cohibin_A.1 | 0.00374 | 0.02508 |
| Coixenolide_2 | 0.00869 | 0.04154 |
| Coixenolide_4 | 0.00561 | 0.03144 |
| Cysteamine_3TMS | 0.00531 | 0.03071 |
| Cysteine_3TMS | 0.01144 | 0.04989 |
| Glycerol-3-phosphate_4TMS.2 | 0.00118 | 0.01276 |
| Glycine | 0.00004 | 0.00173 |
| Guanosine | 0.00001 | 0.00088 |
| Guanosine,_2′-deoxy-_4TMS.1 | 0.00027 | 0.00504 |
| Guanosine_4TMS coeluting_with_Guanosine_5TMS | 0.00261 | 0.02063 |
| Iminodiacetic_acid_3TMS | 0.00185 | 0.01715 |
| Isoleucine_2TMS | 0.00071 | 0.00914 |
| Lactose | 0.00001 | 0.00065 |
| Leucine | 0.00032 | 0.00541 |
| Leucine,_cyclo- | 0.00352 | 0.02428 |
| Leucine_2TMS | 0.00051 | 0.00704 |
| Levulinic_acid | 0.00057 | 0.00760 |
| Luteolin | 0.00474 | 0.02871 |
| Naringenin | 0.00066 | 0.00867 |
| Oxamide_3TMS | 0.00017 | 0.00383 |
| Pantothenic_acid,_D-_3TMS | 0.00799 | 0.04009 |
| Phenylalanine_2TMS | 0.00003 | 0.00147 |
| Phosphomycin | 0.000000008 | 0.00001 |
| Pinitol,_D-_5TMS | 0.00363 | 0.02488 |
| Putrescine_4TMS | 0.00224 | 0.01912 |
| Pyridine | 0.00443 | 0.02729 |
| Pyridoxamine | 0.00859 | 0.04136 |
| Pyroglutamic_acid_2TMS | 0.00976 | 0.04528 |
| Quercetin | 0.00003 | 0.00147 |
| Serine | 0.00000003 | 0.00001 |
| Thiamine | 0.00004 | 0.00173 |
| Threitol,_dithio- | 0.00166 | 0.01589 |
| Thymidine-5′-monophosphoric-acid-3TMS | 0.00020 | 0.00426 |
| Tryptophan_3TMS | 0.00007 | 0.00249 |
| Tyrosine,_2-iodo- | 0.000000017 | 0.00001 |
| Tyrosine_3TMS | 0.00001 | 0.00094 |
| Uric_acid | 0.00017 | 0.00383 |
| Valine_2TMS | 0.00008 | 0.00280 |
| Xanthine_3TMS | 0.00772 | 0.03903 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overton, J.M.; Linke, L.; Magnuson, R.; Broeckling, C.D.; Rao, S. Metabolomic Profiles of Multidrug-Resistant Salmonella Typhimurium from Humans, Bovine, and Porcine Hosts. Animals 2022, 12, 1518. https://doi.org/10.3390/ani12121518
Overton JM, Linke L, Magnuson R, Broeckling CD, Rao S. Metabolomic Profiles of Multidrug-Resistant Salmonella Typhimurium from Humans, Bovine, and Porcine Hosts. Animals. 2022; 12(12):1518. https://doi.org/10.3390/ani12121518
Chicago/Turabian StyleOverton, Jessie M., Lyndsey Linke, Roberta Magnuson, Corey D. Broeckling, and Sangeeta Rao. 2022. "Metabolomic Profiles of Multidrug-Resistant Salmonella Typhimurium from Humans, Bovine, and Porcine Hosts" Animals 12, no. 12: 1518. https://doi.org/10.3390/ani12121518
APA StyleOverton, J. M., Linke, L., Magnuson, R., Broeckling, C. D., & Rao, S. (2022). Metabolomic Profiles of Multidrug-Resistant Salmonella Typhimurium from Humans, Bovine, and Porcine Hosts. Animals, 12(12), 1518. https://doi.org/10.3390/ani12121518