
Analysis of Complete Mitochondrial Genome of Bohadschia argus (Jaeger, 1833) (Aspidochirotida, Holothuriidae)

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Sequence Analysis and Gene Annotation
2.3. Phylogenetic Analysis
3. Results and Discussion
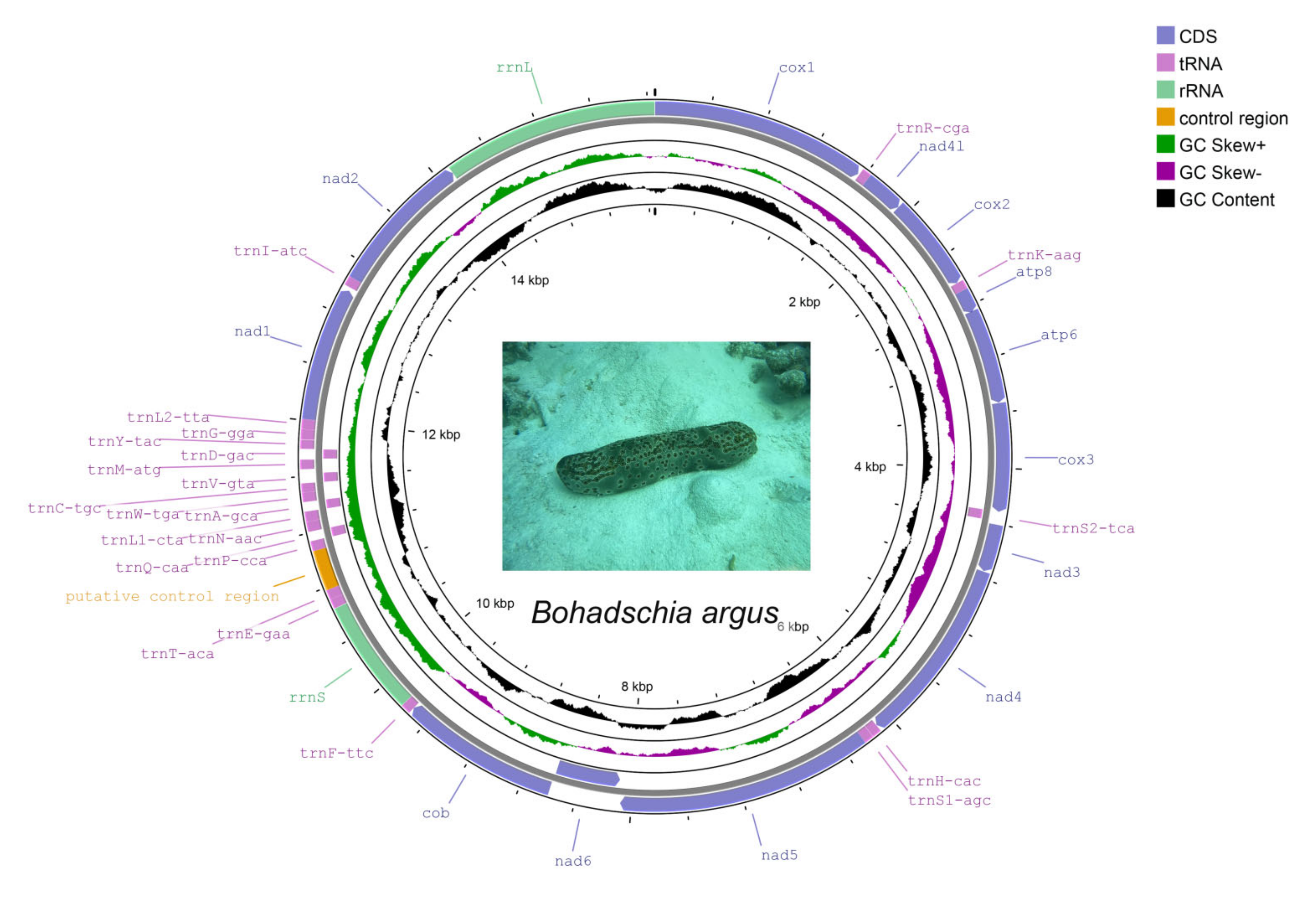
3.1. Mitochondrial Genome Structure and Organization
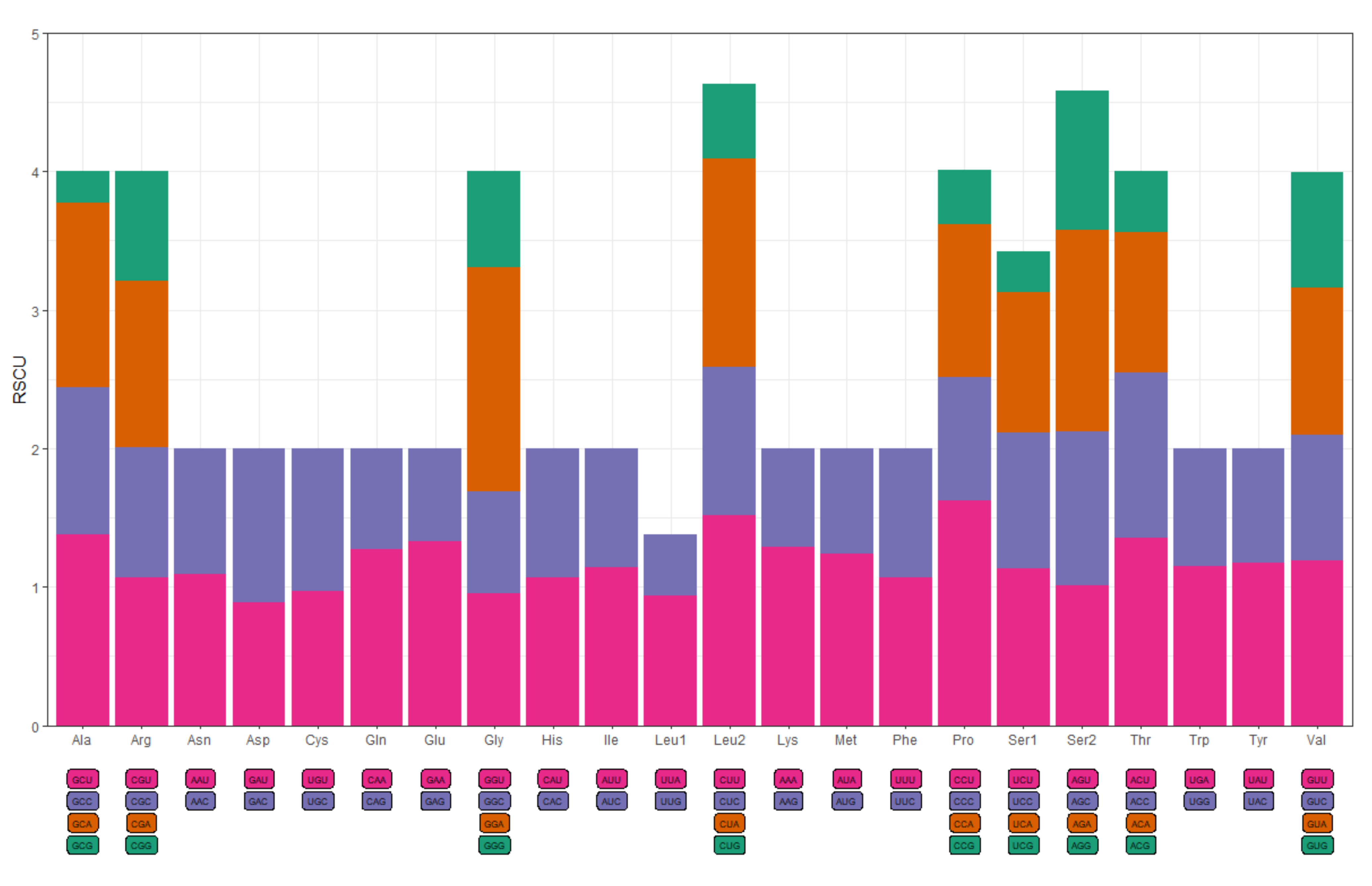
3.2. Protein-Coding Genes
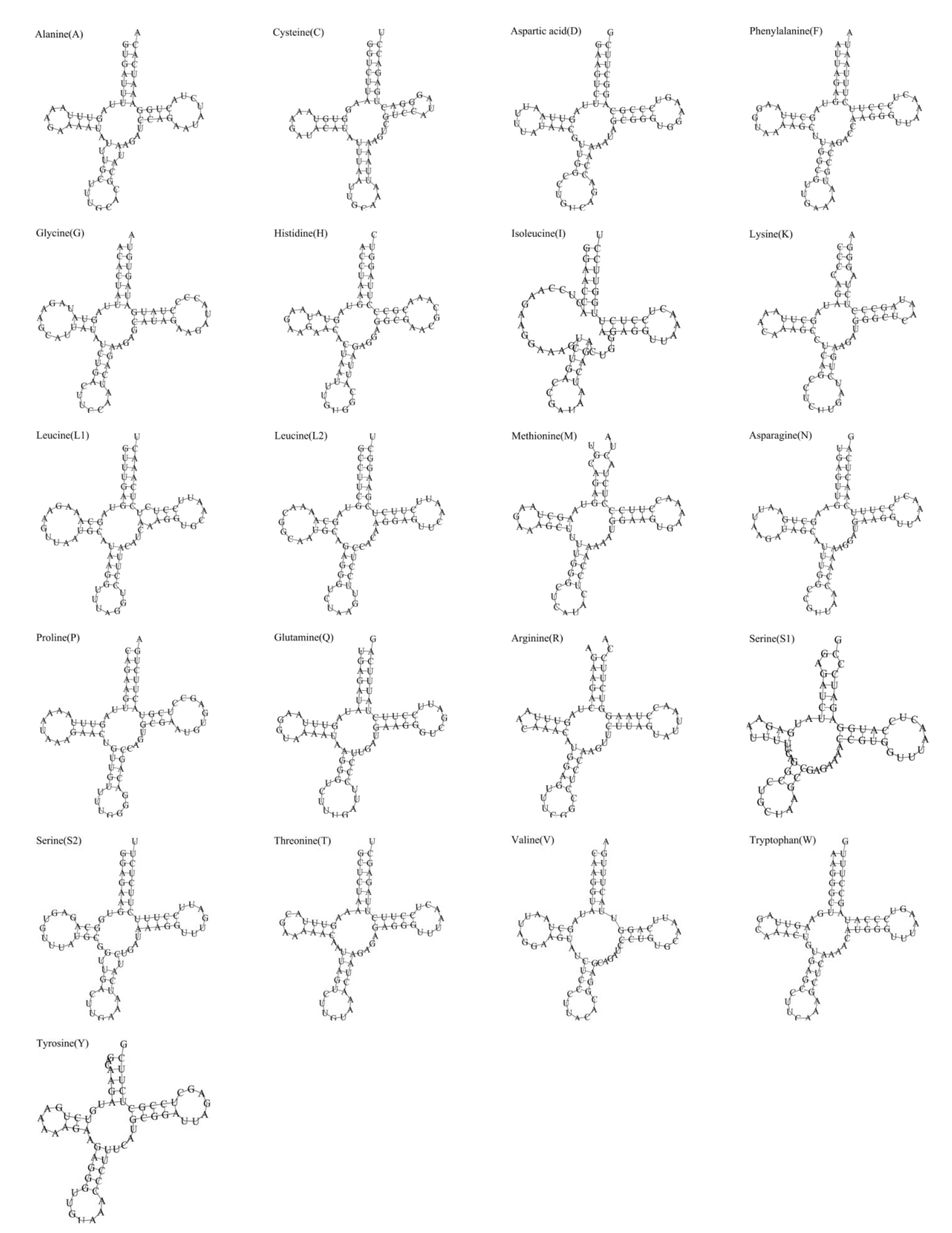
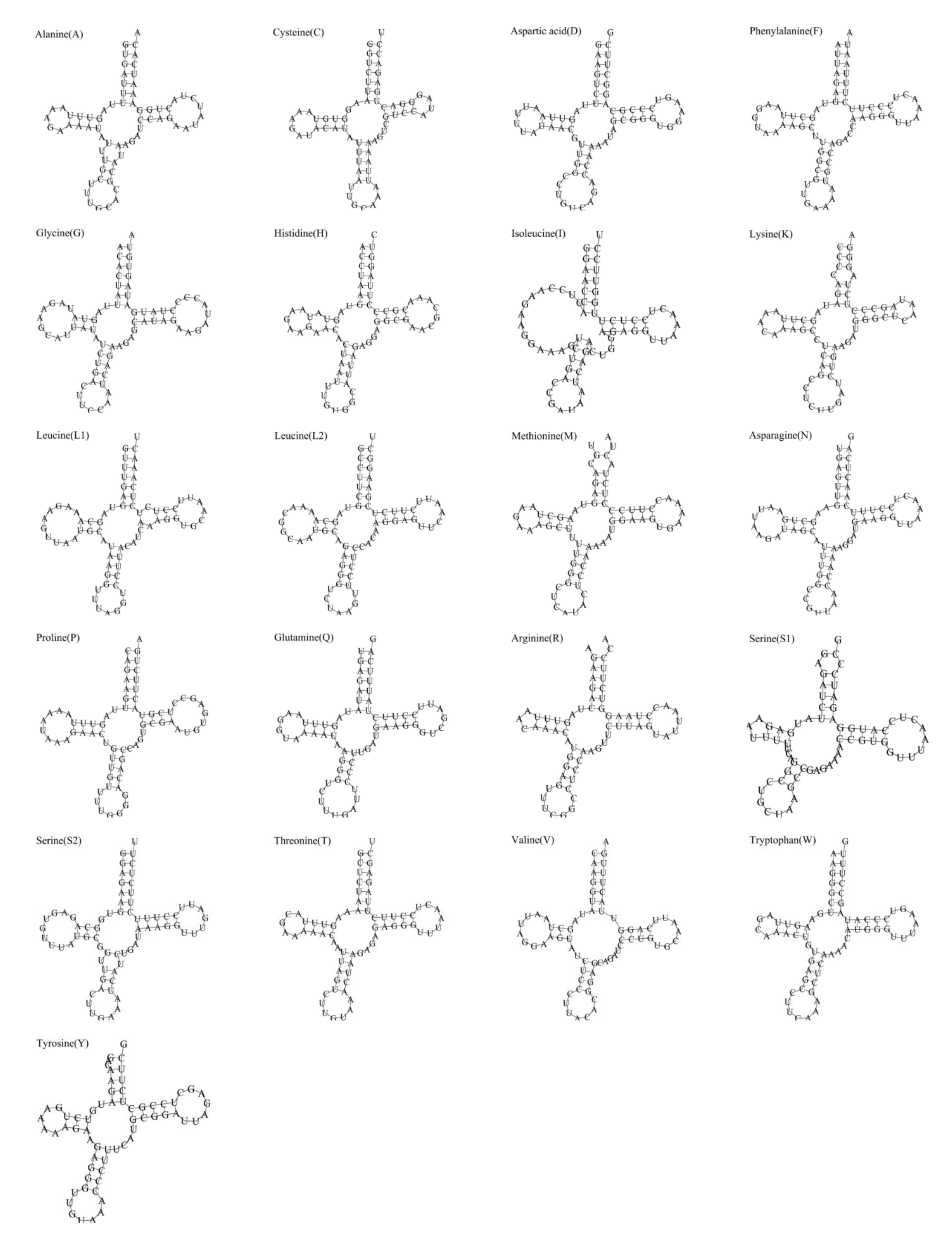
3.3. tRNAs and rRNAs
3.4. Control Region and Overlapping Regions
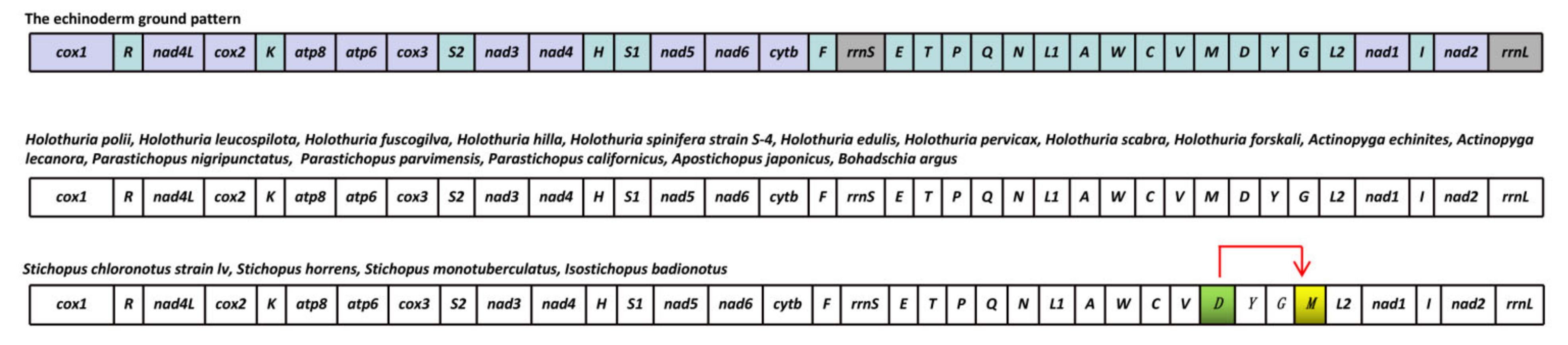
3.5. Gene Arrangement
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Miller, A.K.; Kerr, A.M.; Paulay, G.; Reich, M.; Wilson, N.; Carvajal, J.I.; Rouse, G.W. Molecular phylogeny of extant Holothuroidea (Echinodermata). Mol. Phylogenetics Evol. 2017, 111, 110–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordbar, S.; Anwar, F.; Saari, N. High-Value Components and Bioactives from Sea Cucumbers for Functional Foods—A Review. Mar. Drugs 2011, 9, 1761–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Zhou, C.; Tran, N.T.; Sun, Z.; Wu, J.; Ge, H.; Lu, Z.; Zhong, C.; Zhu, Z.; Yang, Q.; et al. Comparison of the complete mitochondrial genome of Phyllophorus liuwutiensis (Echinodermata: Holothuroidea: Phyllophoridae) to that of other sea cucumbers. FEBS Open Bio 2020, 10, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Hu, C.; Zhang, L.; Fan, S. Genetic identification of global commercial sea cucumber species on the basis of mitochondrial DNA sequences. Food Control 2011, 22, 72–77. [Google Scholar] [CrossRef]
- Honey-Escandón, M.; Laguarda-Figueras, A.; Solís-Marín, F.A. Molecular phylogeny of the subgenus Holothuria (Selenkothuria) Deichmann, 1958 (Holothuroidea: Aspidochirotida). Zool. J. Linn. Soc. 2012, 165, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Kerr, A.M.; Paulay, G. Colour, confusion, and crossing: Resolution of species problems in Bohadschia (Echinodermata: Holothuroidea). Zool. J. Linn. Soc. 2013, 168, 81–97. [Google Scholar] [CrossRef]
- Arndt, A.; Marquez, C.; Lambert, P.; Smith, M.J. Molecular Phylogeny of Eastern Pacific Sea Cucumbers (Echinodermata: Holothuroidea) Based on Mitochondrial DNA Sequence. Mol. Phylogenetics Evol. 1996, 6, 425–437. [Google Scholar] [CrossRef]
- Crispo, E. Modifying effects of phenotypic plasticity on interactions among natural selection, adaptation and gene flow. J. Evol. Biol. 2008, 21, 1460–1469. [Google Scholar] [CrossRef]
- Maan, M.E.; Sefc, K.M. Colour variation in cichlid fish: Developmental mechanisms, selective pressures and evolutionary consequences. Semin. Cell Dev. Biol. 2013, 24, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Gulisija, D.; Kim, Y.; Plotkin, J.B. Phenotypic Plasticity Promotes Balanced Polymorphism in Periodic Environments by a Genomic Storage Effect. Genetics 2016, 202, 1437–1448. [Google Scholar] [CrossRef]
- Pimpinelli, S.; Piacentini, L. Environmental change and the evolution of genomes: Transposable elements as translators of phenotypic plasticity into genotypic variability. Funct. Ecol. 2020, 34, 428–441. [Google Scholar] [CrossRef]
- Solís-Marín, F.A.; Billett, D.S.; Preston, J.; Rogers, A.D. Mitochondrial DNA sequence evidence supporting the recognition of a new North Atlantic Pseudostichopus species (Echinodermata: Holothuroidea). J. Mar. Biol. Assoc. 2004, 84, 1077–1084. [Google Scholar] [CrossRef] [Green Version]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Qiao, Y.; Zhao, L.; Huang, G.; Liu, Y.; Huang, L. Characterization and phylogenetic analysis of the complete mitochondrial genome of Actinopyga lecanora (Jaeger, 1833) (Holothuriida: Holothuriidae). Mitochondrial DNA Part B 2021, 6, 2801–2802. [Google Scholar] [CrossRef]
- Figueroa, A.C.; McHugh, W.J.; Miller, S.M.; Fellgren, A.K.; Bogantes, V.E.; Janosik, A.M. Characterization of the complete mitochondrial genome of Thyonella gemmata (Echinodermata: Cucumariidae). Mitochondrial DNA Part B 2021, 6, 2997–2998. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Zhao, H.; Hu, J.; Chen, B.; Li, H.; Huang, W. Complete mitochondrial genome of a tropical sea cucumber, Stichopus chloronotus. Mitochondrial DNA Part B 2021, 6, 2788–2790. [Google Scholar] [CrossRef]
- Zeng, L.; Wen, J.; Lin, H.; Fan, S.; Sun, Y.; Yang, C.; Zhao, J.; Li, X. The complete mitochondrial genome of Holothuria edulis (Lesson, 1830) (Aspidochirotida, Holothriidae). Mitochondrial DNA Part B 2020, 5, 1471–1472. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.-H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wang, N.; Tan, J.; Wang, R.; Yang, Y.; Li, G.; Guan, H.; Zheng, Y.; Shi, X.; Ye, R.; et al. Comprehensive codon usage analysis of porcine deltacoronavirus. Mol. Phylogenetics Evol. 2019, 141, 106618. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Bernt, M.; Merkle, D.; Ramsch, K.; Fritzsch, G.; Perseke, M.; Bernhard, D.; Schlegel, M.; Stadler, P.F.; Middendorf, M. CREx: Inferring genomic rearrangements based on common intervals. Bioinformatics 2007, 23, 2957–2958. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Sha, Z.; Xiao, N. The first two complete mitogenomes of the order Apodida from deep-sea chemoautotrophic environments: New insights into the gene rearrangement, origin and evolution of the deep-sea sea cucumbers. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 39, 100839. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kazutaka, K.; Misakwa, K.; Kei-ichi, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Liu, Y.; Huang, L.; Zhao, Y.; Huang, G. The complete mitochondrial genome of black sea cucumber Holothuria leucospilota (Aspidochirotida holothuriidae). Mitochondrial DNA Part B 2019, 4, 3377–3378. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Sun, Y.; Rong, F.; Guo, C.; Wang, Y.; Zhang, Y.; Gao, P.; Ding, J. The complete mitochondrial genome of Holothuria spinifera (Théel, 1866). Mitochondrial DNA Part B 2020, 5, 1679–1680. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Ding, B.; Guo, C.; Han, R.; Li, J.; Rong, F.; Ding, J. The complete mitochondrial genome of Holothuria fuscocinerea (Jaeger,1833). Mitochondrial DNA Part B 2019, 5, 33–34. [Google Scholar] [CrossRef] [Green Version]
- Utzeri, V.J.; Ribani, A.; Bovo, S.; Taurisano, V.; Calassanzio, M.; Baldo, D.; Fontanesi, L. Microscopic ossicle analyses and the complete mitochondrial genome sequence of Holothuria (Roweothuria) polii (Echinodermata; Holothuroidea) provide new information to support the phylogenetic positioning of this sea cucumber species. Mar. Genom. 2020, 51, 100735. [Google Scholar] [CrossRef]
- Kono, N.; Tomita, M.; Arakawa, K. Accelerated Laboratory Evolution Reveals the Influence of Replication on the GC Skew in Escherichia coli. Genome Biol. Evol. 2018, 10, 3110–3117. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Bae, Y.J.; Shin, S. Mitochondrial gene rearrangement and phylogenetic relationships in the Amphilepidida and Ophiacanthida (Echinodermata, Ophiuroidea). Mar. Biol. Res. 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Mu, W.; Liu, J.; Zhang, H. Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS ONE 2018, 13, e0208051. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, X.; Tao, Y.; Wu, Q.; Bai, Y.; Guo, H.; Tang, D. The complete mitochondrial genome of Parasesarma pictum (Brachyura: Grapsoidea: Sesarmidae) and comparison with other Brachyuran crabs. Genomics 2019, 111, 799–807. [Google Scholar] [CrossRef]
- Liu, Q.N.; Zhu, B.J.; Dai, L.S.; Wei, G.Q.; Liu, C.L. The Complete Mitochondrial Genome of the Wild Silkworm Moth, Actias Selene. Gene 2012, 505, 291–299. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. Transfer-RNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Ivey, J.L.; Santos, S.R. The complete mitochondrial genome of the Hawaiian anchialine shrimp Halocaridina rubra Holthuis, 1963 (Crustacea: Decapoda: Atyidae). Gene 2007, 394, 35–44. [Google Scholar] [CrossRef]
- Xin, Y.; Ren, J.; Liu, X. Mitogenome of the small abalone Haliotis diversicolor Reeve and phylogenetic analysis within Gastropoda. Mar. Genom. 2011, 4, 253–262. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Jiang, F.; Guo, X.; Liu, B. Unusual conservation of mitochondrial gene order in Crassostreaoysters: Evidence for recent speciation in Asia. BMC Evol. Biol. 2010, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Shen, X.; Jiang, F.; Liu, B. The Mitochondrial Genomes of Two Scallops, Argopecten irradians and Chlamys farreri (Mollusca: Bivalvia): The Most Highly Rearranged Gene Order in the Family Pectinidae. J. Mol. Evol. 2010, 70, 57–68. [Google Scholar] [CrossRef]
- Xia, J.; Ren, C.; Yu, Z.; Wu, X.; Qian, J.; Hu, C. Complete mitochondrial genome of the sandfish Holothuria scabra (Holothuroidea, Holothuriidae). Mitochondrial DNA Part A 2016, 27, 4174–4175. [Google Scholar] [CrossRef]
- Fourdrilis, S.; Martins, A.M.D.F.; Backeljau, T. Relation between mitochondrial DNA hyperdiversity, mutation rate and mitochondrial genome evolution in Melarhaphe neritoides (Gastropoda: Littorinidae) and other Caenogastropoda. Sci. Rep. 2018, 8, 17964. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.-I.; Suematsu, T.; Ohtsuki, T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front. Genet. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, G.; McClain, W.H. The G·U wobble base pair—A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 2000, 1, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.R. Animal Mitochondrial DNA: Structure and Evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [CrossRef]
- Arndt, A.; Smith, M.J. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol. Biol. Evol. 1998, 15, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Scouras, A.; Beckenbach, K.; Arndt, A.; Smith, M.J. Complete mitochondrial genome DNA sequence for two ophiuroids and a holothuroid: The utility of protein gene sequence and gene maps in the analyses of deep deuterostome phylogeny. Mol. Phylogenetics Evol. 2004, 31, 50–65. [Google Scholar] [CrossRef]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Shen, X.; Tian, M.; Liu, Z.; Cheng, H.; Tan, J.; Meng, X.; Ren, J. Complete mitochondrial genome of the sea cucumber Apostichopus japonicus (Echinodermata: Holothuroidea): The first representative from the subclass Aspidochirotacea with the echinoderm ground pattern. Gene 2009, 439, 79–86. [Google Scholar] [CrossRef]
- Li, Z.; Ma, B.; Li, X.; Lv, Y.; Jiang, X.; Ren, C.; Hu, C.; Luo, P. The Complete Mitochondrial Genome of Stichopus naso (Aspidochirotida: Stichopodidae: Stichopus) and Its Phylogenetic Position. Genes 2022, 13, 825. [Google Scholar] [CrossRef]
- Limei, C.; Qi, L.I.; Yun, L.I. Sequence analysis of mitochondrial 16S rRNA and CO1 gene and molecular phylogeny of four species of sea cucumber. J. Fish. Sci. China 2008, 15, 935–942. (In Chinese) [Google Scholar]
- Lacey, K.M.J.; McCormack, G.P.; Keegan, B.F.; Powell, R. Phylogenetic relationships within the class holothuroidea, inferred from 18S rRNA gene data. Mar. Biol. 2005, 147, 1149–1154. [Google Scholar] [CrossRef]
- VandenSpiegel, D.; Jangoux, M. Fine Structure and Behaviour of the So-called Cuvierian Organs in the Holothuroid GenusActinopyga (Echinodermata). Acta Zool. 1993, 74, 43–50. [Google Scholar] [CrossRef]
- Pearson, J. Proposed re-classification of the genera Mülleria and Holothuria. Spolia Zeylanica 1914, 9, 163–172. [Google Scholar] [CrossRef]
- Kerr, A.M.; Janies, D.A.; Clouse, R.M.; Samyn, Y.; Kuszak, J.; Kim, J. Molecular Phylogeny of Coral-Reef Sea Cucumbers (Holothuriidae: Aspidochirotida) Based on 16S Mitochondrial Ribosomal DNA Sequence. Mar. Biotechnol. 2005, 7, 53–60. [Google Scholar] [CrossRef]
- Zhong, S.; Huang, L.; Liu, Y.; Huang, G. The first complete mitochondrial genome of Actinopyga from Actinopyga echinites (Aspidochirotida: Holothuriidae). Mitochondrial DNA Part B 2020, 5, 854–855. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Species | Sizes (bp) | Accession No. |
|---|---|---|---|
| Holothuriidae | Holothuria polii | 15,907 | LR694133.1 |
| Holothuriidae | Holothuria leucospilota | 15,904 | NC_046849.1 |
| Holothuriidae | Holothuria fuscogilva | 15,633 | MZ305460.1 |
| Holothuriidae | Holothuria hilla | 15,744 | MN163001.1 |
| Holothuriidae | Holothuria spinifera strain S-4 | 15,812 | NC_046508.1 |
| Holothuriidae | Holothuria edulis | 15,743 | NC_051928.1 |
| Holothuriidae | Holothuria fuscocinerea | 15,633 | MZ305460.1 |
| Holothuriidae | Holothuria scabra | 15,779 | NC_027086.1 |
| Holothuriidae | Holothuria forskali | 15,841 | NC_013884.1 |
| Holothuriidae | Actinopyga echinites | 15,619 | MN793975.1 |
| Holothuriidae | Actinopyga lecanora | 15,568 | MW248463.1 |
| Stichopodidae | Stichopus monotuberculatus | 16,274 | NC_052743.1 |
| Stichopodidae | Stichopus chloronotus strain lv | 16,247 | NC_056131.1 |
| Stichopodidae | Stichopus horrens | 16,257 | NC_014454.1 |
| Stichopodidae | Parastichopus nigripunctatus | 16,112 | NC_013432.1 |
| Stichopodidae | Isostichopus badionotus | 16,318 | MZ188901.1 |
| Stichopodidae | Parastichopus parvimensis | 16,120 | NC_029699.1 |
| Stichopodidae | Parastichopus californicus | 16,727 | NC_026727.1 |
| Stichopodidae | Apostichopus japonicus | 16,099 | NC_012616.1 |
| Psychropotidae | Benthodytes | 17,567 | NC_040968.1 |
| Elpidiidae | Scotoplanes | 15,910 | LC416626.1 |
| Phyllophoridae | Phyllophorella liuwutiensis | 15,969 | NC_057437.1 |
| Phyllophoridae | Phyrella fragilis | 15,910 | MZ305459.1 |
| Cucumariidae | Thyonella gemmata voucher UF:021831 | 15,696 | MZ463652.1 |
| Cucumariidae | Neocucumis proteus | 16,495 | MZ305458.1 |
| Cucumariidae | Cercodemas anceps | 16,539 | NC_054245.1 |
| Cucumariidae | Pseudocolochirus violaceus | 15,756 | NC_051967 |
| Cucumariidae | Colochirus quadrangularis | 17,157 | NC_051929.1 |
| Cucumariidae | Cucumaria miniata | 17,538 | NC_005929.1 |
| Chiridotidae | Chiridota sp. SS-2021 | 17,199 | MW357262.1 |
| Chiridotidae | Chiridota heheva | 17,200 | MW357261.1 |
| B. argus | Size | A% | T% | G% | C% | AT% | GC% | AT-Skew | GC-Skew |
|---|---|---|---|---|---|---|---|---|---|
| mitogenome | 15,656 | 32.12 | 26.9 | 17.64 | 23.35 | 59.01 | 40.99 | 0.09 | −0.14 |
| PCGs | 11,354 | 29.97 | 28.93 | 17.08 | 24.02 | 58.90 | 41.10 | 0.02 | −0.17 |
| tRNAs | 1525 | 31.02 | 27.8 | 22.23 | 18.95 | 58.82 | 41.18 | 0.05 | 0.08 |
| rRNAs | 2371 | 37.24 | 21.09 | 20.29 | 21.38 | 58.33 | 41.67 | 0.28 | −0.03 |
| control region | 290 | 22.41 | 38.28 | 16.55 | 22.76 | 60.69 | 39.31 | −0.26 | −0.16 |
| Gene | Direction | Location | Size | Start Codon | Stop Codon | Anticodon |
|---|---|---|---|---|---|---|
| cox1 | + | 1–1554 | 1554 | ATG | TAG | |
| trnR | + | 1564–1630 | 67 | CGA | ||
| nad4l | + | 1631–1927 | 297 | ATG | TAA | |
| cox2 | + | 1929–2616 | 688 | ATG | T | |
| trnK | + | 2617–2682 | 66 | AAG | ||
| atp8 | + | 2683–2850 | 168 | ATG | TAA | |
| atp6 | + | 2844–3527 | 684 | ATG | TAA | |
| cox3 | + | 3530–4312 | 783 | ATG | TAA | |
| trnS2 | − | 4312–4382 | 71 | TCA | ||
| nad3 | + | 4405–4749 | 345 | ATG | TAA | |
| nad4 | + | 4753–6109 | 1357 | ATG | T | |
| trnH | + | 6111–6178 | 68 | CAC | ||
| trnS1 | + | 6180–6247 | 68 | AGC | ||
| nad5 | + | 6248–8080 | 1833 | ATG | TAG | |
| nad6 | − | 8097–8585 | 489 | ATG | TAA | |
| cob | + | 8594–9736 | 1143 | ATG | TAA | |
| trnF | + | 9736–9806 | 71 | TTC | ||
| rrnS | + | 9808–10,637 | 830 | |||
| trnE | + | 10,640–10,709 | 70 | GAA | ||
| trnT | + | 10,710–10,779 | 70 | ACA | ||
| trnP | + | 11,070–11,141 | 72 | CCA | ||
| trnQ | − | 11,138–11,207 | 70 | CAA | ||
| trnN | + | 11,209–11,281 | 73 | AAC | ||
| trnL1 | + | 11,282–11,353 | 72 | CTA | ||
| trnA | − | 11,353–11,420 | 68 | GCA | ||
| trnW | + | 11,421–11,488 | 68 | TGA | ||
| trnC | + | 11,489–11,553 | 65 | TGC | ||
| trnV | − | 11,553–11,622 | 70 | GTA | ||
| trnM | + | 11,653–11,721 | 69 | ATG | ||
| trnD | − | 11,727–11,796 | 70 | GAC | ||
| trnY | + | 11,797–11,861 | 65 | TAC | ||
| trnG | + | 11,864–11,936 | 73 | GGA | ||
| trnL2 | + | 11,937–12,007 | 71 | TTA | ||
| nad1 | + | 12,008–12,979 | 972 | ATG | TAA | |
| trnI | + | 13,003–13070 | 68 | ATC | ||
| nad2 | + | 13071–14,111 | 1041 | ATG | TAA | |
| rrnL | + | 14,112–15,652 | 1541 | |||
| Controlregion | 10,780–11,069 | 290 |
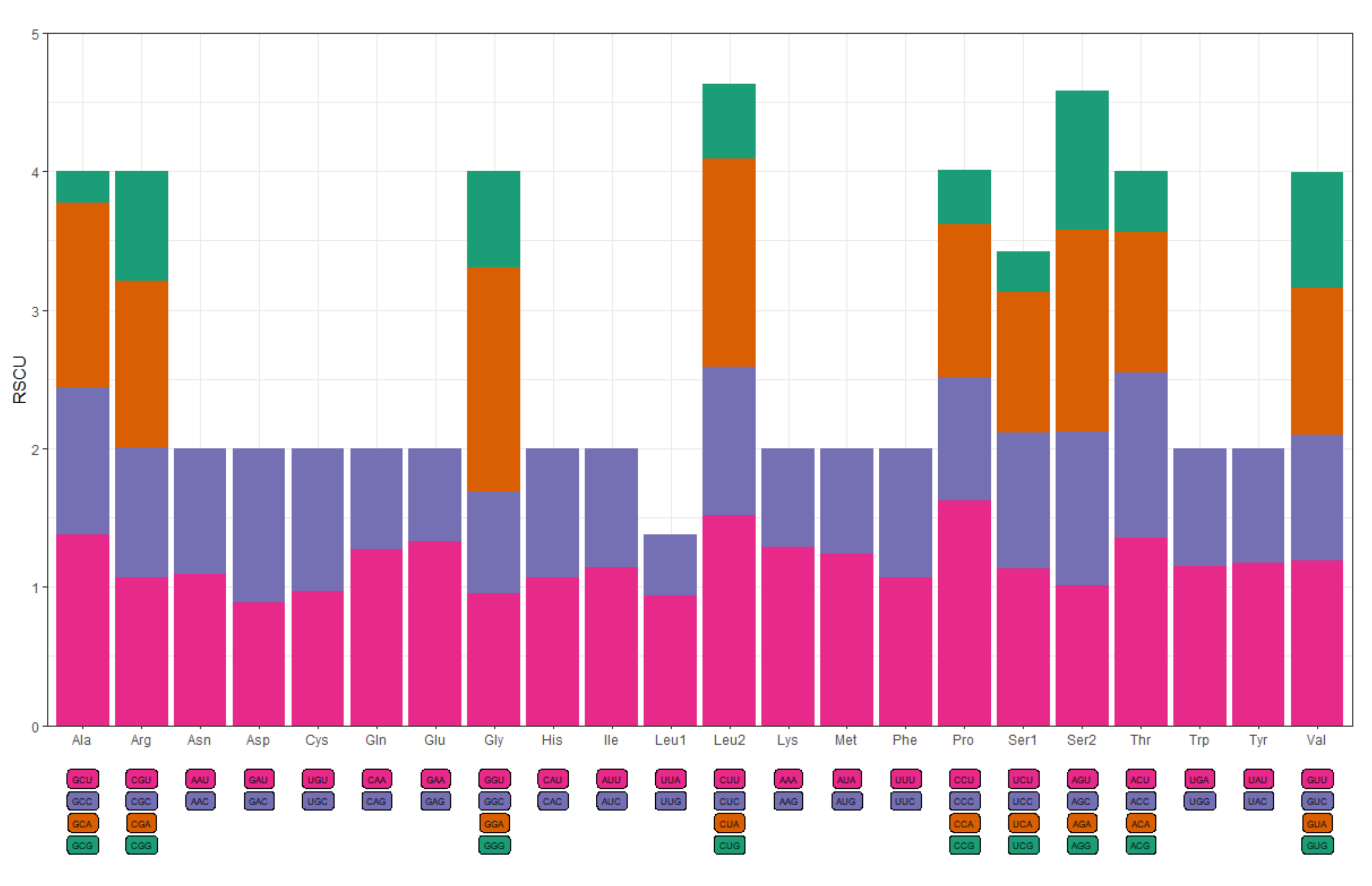
| Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|
| UUU(F) | 137 | 1.07 | UCU(S) | 103 | 1.13 |
| UUC(F) | 119 | 0.93 | UCC(S) | 89 | 0.98 |
| UUA(L) | 84 | 0.94 | UCA(S) | 93 | 1.02 |
| UUG(L) | 39 | 0.44 | UCG(S) | 26 | 0.29 |
| CUU(L) | 136 | 1.52 | CCU(P) | 146 | 1.62 |
| CUC(L) | 96 | 1.07 | CCC(P) | 80 | 0.89 |
| CUA(L) | 134 | 1.50 | CCA(P) | 100 | 1.11 |
| CUG(L) | 48 | 0.54 | CCG(P) | 35 | 0.39 |
| UAU(Y) | 119 | 1.17 | AUU(I) | 108 | 1.14 |
| UAC(Y) | 84 | 0.83 | AUC(I) | 81 | 0.86 |
| UAA(*) | 125 | 1.24 | AUA(M) | 145 | 1.24 |
| UAG(*) | 77 | 0.76 | AUG(M) | 88 | 0.76 |
| CAU(H) | 78 | 1.07 | GUU(V) | 56 | 1.19 |
| CAC(H) | 68 | 0.93 | GUC(V) | 43 | 0.91 |
| CAA(Q) | 94 | 1.27 | GUA(V) | 50 | 1.06 |
| CAG(Q) | 54 | 0.73 | GUG(V) | 39 | 0.83 |
| ACU(T) | 117 | 1.35 | AAU(N) | 140 | 1.09 |
| ACC(T) | 104 | 1.20 | AAC(N) | 118 | 0.91 |
| ACA(T) | 87 | 1.01 | AAA(K) | 217 | 1.29 |
| ACG(T) | 38 | 0.44 | AAG(K) | 119 | 0.71 |
| GCU(A) | 72 | 1.38 | GAU(D) | 60 | 0.89 |
| GCC(A) | 55 | 1.06 | GAC(D) | 75 | 1.11 |
| GCA(A) | 69 | 1.33 | GAA(E) | 114 | 1.33 |
| GCG(A) | 12 | 0.23 | GAG(E) | 58 | 0.67 |
| UGU(C) | 43 | 0.97 | CGU(R) | 34 | 1.07 |
| UGC(C) | 46 | 1.03 | CGC(R) | 30 | 0.94 |
| UGA(W) | 74 | 1.15 | CGA(R) | 38 | 1.20 |
| UGG(W) | 55 | 0.85 | CGG(R) | 25 | 0.79 |
| AGU(S) | 92 | 1.01 | GGU(G) | 54 | 0.95 |
| AGC(S) | 101 | 1.11 | GGC(G) | 42 | 0.74 |
| AGA(S) | 133 | 1.46 | GGA(G) | 92 | 1.62 |
| AGG(S) | 91 | 1.00 | GGG(G) | 39 | 0.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, B.; Li, Z.; Lv, Y.; E, Z.; Fang, J.; Ren, C.; Luo, P.; Hu, C. Analysis of Complete Mitochondrial Genome of Bohadschia argus (Jaeger, 1833) (Aspidochirotida, Holothuriidae). Animals 2022, 12, 1437. https://doi.org/10.3390/ani12111437
Ma B, Li Z, Lv Y, E Z, Fang J, Ren C, Luo P, Hu C. Analysis of Complete Mitochondrial Genome of Bohadschia argus (Jaeger, 1833) (Aspidochirotida, Holothuriidae). Animals. 2022; 12(11):1437. https://doi.org/10.3390/ani12111437
Chicago/Turabian StyleMa, Bo, Zhuobo Li, Ying Lv, Zixuan E, Jianxiang Fang, Chunhua Ren, Peng Luo, and Chaoqun Hu. 2022. "Analysis of Complete Mitochondrial Genome of Bohadschia argus (Jaeger, 1833) (Aspidochirotida, Holothuriidae)" Animals 12, no. 11: 1437. https://doi.org/10.3390/ani12111437
APA StyleMa, B., Li, Z., Lv, Y., E, Z., Fang, J., Ren, C., Luo, P., & Hu, C. (2022). Analysis of Complete Mitochondrial Genome of Bohadschia argus (Jaeger, 1833) (Aspidochirotida, Holothuriidae). Animals, 12(11), 1437. https://doi.org/10.3390/ani12111437