
Microbiota Composition of Mucosa and Interactions between the Microbes of the Different Gut Segments Could Be a Factor to Modulate the Growth Rate of Broiler Chickens

 ,
,  , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics and Approval Statement
2.2. Animal Handling and Sampling
2.3. DNA Extraction, 16S rRNA Gene Amplification and Illumina MiSeq Sequencing
2.4. Bioinformatics and Statistical Analyses
2.5. Co-Occurrence Network Analysis
3. Results
3.1. Sequencing Data and Differences in the Alpha and Beta Diversity
3.2. Gut Microbiota Composition at Phylum Level
3.3. Gut Microbiota Composition at Genus Level
3.4. Correlation between Gut Microbiota and BW
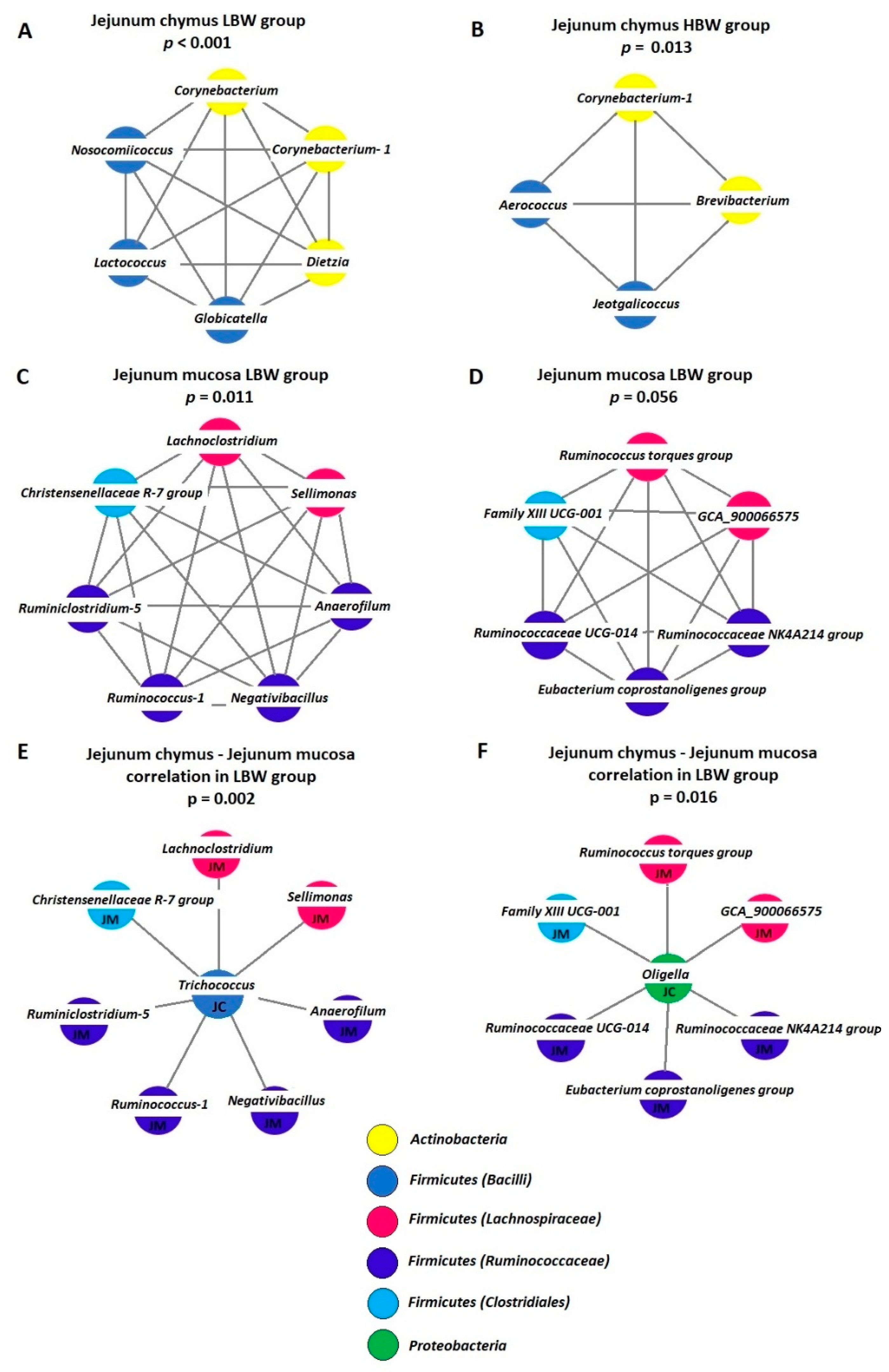
3.5. Analysis of Co-Occurrence Patterns
4. Discussion
4.1. Diversity of Gut Microbiota
4.2. Composition of Gut Microbiota in the Different Sampling Places
4.3. BW-Related Differences of the Gut Microbiota
4.4. Analysis of Co-Occurrence Patterns
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stanley, D.; Denman, S.E.; Hughes, R.J.; Geier, M.S.; Crowley, T.M.; Chen, H.; Haring, V.R.; Moore, R.J. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biotechnol. 2012, 96, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Sun, C.; Yuan, J.; Yang, N. Gut metagenomic analysis reveals prominent roles of Lactobacillus and cecal microbiota in chicken feed efficiency. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Torok, V.A.; Allison, G.E.; Percy, N.J.; Ophel-Keller, K.; Hughes, R.J. Influence of antimicrobial feed additives on broiler commensal posthatch gut microbiota development and performance. Appl. Environ. Microbiol. 2011, 77, 3380–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, K.M.; Shah, T.; Deshpande, S.; Jakhesara, S.J.; Koringa, P.G.; Rank, D.N.; Joshi, C.G. High through put 16S rRNA gene-based pyrosequencing analysis of the fecal microbiota of high FCR and low FCR broiler growers. Mol. Biol. Rep. 2012, 39, 10595–10602. [Google Scholar] [CrossRef]
- Liu, J.; Stewart, S.N.; Robinson, K.; Yang, Q.; Lyu, W.; Whitmore, M.A.; Zhang, G. Linkage between the intestinal microbiota and residual feed intake in broiler chickens. J. Anim. Sci. Biotechnol. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Gou, Y.K.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Salaheen, S.; Kim, S.-W.; Haley, B.J.; Van Kessel, J.A.S.; Biswas, D. Alternative Growth Promoters Modulate Broiler Gut Microbiome and Enhance Body Weight Gain. Front. Microbiol. 2017, 8, 2088. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Lobley, G.E.; Holtrop, G.; Ince, J.; Johnstone, A.M.; Louis, P.; Flint, H.J. Human colonic microbiota associated with diet, obesity and weight loss. Int. J. Obes. 2008, 32, 1720–1724. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ppatil, D.; Pdhotre, D.; Gchavan, S.; Sultan, A.; Jain, D.S.; Lanjekar, V.B.; Gangawani, J.; Sshah, P.; Stodkar, J.; Shah, S.; et al. Molecular analysis of gut microbiota in obesity among Indian individuals. J. Biosci. 2012, 37, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef] [PubMed]
- Medvecky, M.; Cejkova, D.; Polansky, O.; Karasova, D.; Kubasova, T.; Rychlik, I.; Cizek, A. Whole genome sequencing and function prediction of 133 gut anaerobes isolated from chicken caecum in pure cultures. BMC Genomics 2018, 19, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borda-Molina, D.; Seifert, J.; Camarinha-Silva, A. Current Perspectives of the Chicken Gastrointestinal Tract and Its Microbiome. Comput. Struct. Biotechnol. J. 2018, 16, 131–139. [Google Scholar] [CrossRef]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial Co-occurrence Relationships in the Human Microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Aviagen. Ross Brioler Management Handbook; Aviagen Group, Cummings Research Park, 920 Explorer Boulevard NW: Huntsville, AL, USA, 2018. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewski, M.; Proietti, C.; Ellis, J.J.; Hasan, S.; Brion, M.J.; Berger, B.; Krause, L. Calypso: A user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 2017, 33, 782–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanca, M.; Alarcón, R.; Arnau, J.; Bono, R.; Bendayan, R. Non-normal data: Is ANOVA still a valid option? Psicothema 2017, 29, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef]
- Stanley, D.; Geier, M.S.; Hughes, R.J.; Denman, S.E.; Moore, R.J. Highly variable microbiota development in the chicken gastrointestinal tract. PLoS ONE 2013, 8, e84290. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Lin, L.; Zheng, L.; Tang, H.; Fan, X.; Xue, N.; Li, M.; Liu, M.; Li, X. Cecal microbiome profile altered by Salmonella enterica, serovar Enteritidis inoculation in chicken. Gut Pathog. 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Siegerstetter, S.-C.; Schmitz-Esser, S.; Magowan, E.; Wetzels, S.U.; Zebeli, Q.; Lawlor, P.G.; O’Connell, N.E.; Metzler-Zebeli, B.U. Intestinal microbiota profiles associated with low and high residual feed intake in chickens across two geographical locations. PLoS ONE 2017, 12, e0187766. [Google Scholar] [CrossRef]
- Bae, Y.; Koo, B.; Lee, S.; Mo, J.; Oh, K.; Mo, I.P. Bacterial diversity and its relationship to growth performance of broilers. Korean J. Vet. Res. 2017, 57, 159–167. [Google Scholar] [CrossRef]
- Borda-Molina, D.; Vital, M.; Sommerfeld, V.; Rodehutscord, M.; Camarinha-Silva, A. Insights into broilers’ gut microbiota fed with phosphorus, calcium, and phytase supplemented diets. Front. Microbiol. 2016, 7, 2033. [Google Scholar] [CrossRef] [Green Version]
- Han, G.G.; Kim, E.B.; Lee, J.; Lee, J.Y.; Jin, G.; Park, J.; Huh, C.S.; Kwon, I.K.; Kil, D.Y.; Choi, Y.J.; et al. Relationship between the microbiota in different sections of the gastrointestinal tract, and the body weight of broiler chickens. Springerplus 2016, 5, 911. [Google Scholar] [CrossRef] [Green Version]
- Ranjitkar, S.; Engberg, R.; Lawley, B.; Tannock, G. Bacterial succession in the broiler gastrointestinal tract. Appl. Environ. Microbiol. 2016, 82, 2399–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glendinning, L.; Watson, K.A.; Watson, M. Development of the duodenal, ileal, jejunal and caecal microbiota in chickens. Anim. Microbiome 2019, 1, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Sekelja, M.; Rud, I.; Knutsen, S.H.; Denstadli, V.; Westereng, B.; Næs, T.; Rudi, K. Abrupt temporal fluctuations in the chicken fecal microbiota are explained by its gastrointestinal origin. Appl. Environ. Microbiol. 2012, 78, 2941–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Videnska, P.; Faldynova, M.; Juricova, H.; Babak, V.; Sisak, F.; Havlickova, H.; Rychlik, I. Chicken faecal microbiota and disturbances induced by single or repeated therapy with tetracycline and streptomycin. BMC Vet. Res. 2013, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Rychlik, I. Composition and Function of Chicken Gut Microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Witzig, M.; Da Silva, A.C.; Green-Engert, R.; Hoelzle, K.; Zeller, E.; Seifert, J.; Hoelzle, L.E.; Rodehutscord, M. Spatial Variation of the Gut Microbiota in Broiler Chickens as Affected by Dietary Available Phosphorus and Assessed by T-RFLP Analysis and 454 Pyrosequencing. PLoS ONE 2015, 10, e0143442. [Google Scholar] [CrossRef]
- Stanley, D.; Hughes, R.J.; Moore, R.J. Microbiota of the chicken gastrointestinal tract: Influence on health, productivity and disease. Appl. Microbiol. Biotechnol. 2014, 98, 4301–4310. [Google Scholar] [CrossRef]
- Awad, W.A.; Mann, E.; Dzieciol, M.; Hess, C.; Schmitz-Esser, S.; Wagner, M.; Hess, M. Age-Related Differences in the Luminal and Mucosa-Associated Gut Microbiome of Broiler Chickens and Shifts Associated with Campylobacter jejuni Infection. Front. Cell. Infect. Microbiol. 2016, 6, 154. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Xiang, Y.; Zhou, W.; Chen, J.; Li, K.; Yang, H. Microbial community mapping in intestinal tract of broiler chicken. Poult. Sci. 2017, 96, 1387–1393. [Google Scholar] [CrossRef]
- Mohd Shaufi, M.A.; Sieo, C.C.; Chong, C.W.; Gan, H.M.; Ho, Y.W. Deciphering chicken gut microbial dynamics based on high-throughput 16S rRNA metagenomics analyses. Gut Pathog. 2015, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Idris, U.; Harmon, B.; Hofacre, C.; Maurer, J.J.; Lee, M.D. Diversity and Succession of the Intestinal Bacterial Community of the Maturing Broiler Chicken. Appl. Environ. Microbiol. 2003, 69, 6816–6824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehata, A.A.; Schrödl, W.; Aldin, A.A.; Hafez, H.M.; Krüger, M. The Effect of Glyphosate on Potential Pathogens and Beneficial Members of Poultry Microbiota In Vitro. Curr. Microbiol. 2012, 66, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, M.; Lu, Y.; Guo, X.; Qiao, F.; Wu, L. Antibacterial and residual antimicrobial activities against Enterococcus faecalis biofilm: A comparison between EDTA, chlorhexidine, cetrimide, MTAD and QMix. Sci. Rep. 2015, 5, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and inflammation. Cell 2014, 157, 121. [Google Scholar] [CrossRef] [Green Version]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and Functional Features of the Gastrointestinal Microbiome and Their Effects on Human Health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Adewole, D.; Akinyemi, F. Gut microbiota dynamics, growth performance, and gut morphology in broiler chickens fed diets varying in energy density with or without bacitracin methylene disalicylate (Bmd). Microorganisms 2021, 9, 787. [Google Scholar] [CrossRef]
- Gosalbes, M.J.; Vázquez-Castellanos, J.F.; Angebault, C.; Woerther, P.L.; Ruppé, E.; Ferrús, M.L.; Latorre, A.; Andremont, A.; Moya, A. Carriage of Enterobacteria Producing Extended-Spectrum β-Lactamases and Composition of the Gut Microbiota in an Amerindian Community. Antimicrob. Agents Chemother. 2015, 60, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Li, D.; Zhou, B.; Jiang, Y.; Bai, H.; Zhang, Y.; Xu, Q.; Zhao, W.; Chen, G. Comparative characterization of bacterial communities in geese consuming of different proportions of ryegrass. PLoS ONE 2019, 14, e0223445. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezeji, J.C.; Sarikonda, D.K.; Hopperton, A.; Erkkila, H.L.; Cohen, D.E.; Martinez, S.P.; Cominelli, F.; Kuwahara, T.; Dichosa, A.E.K.; Good, C.E.; et al. Parabacteroides distasonis: Intriguing aerotolerant gut anaerobe with emerging antimicrobial resistance and pathogenic and probiotic roles in human health. Gut Microbes 2021, 13, 1922241. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.J.; Wearsch, P.A.; Veloo, A.C.M.; Rodriguez-Palacios, A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front. Immunol. 2020, 11, 906. [Google Scholar] [CrossRef] [PubMed]
- Duggett, N.A.; Kay, G.L.; Sergeant, M.J.; Bedford, M.; Constantinidou, C.I.; Penn, C.W.; Millard, A.D.; Pallen, M.J. Draft Genome Sequences of Six Novel Bacterial Isolates from Chicken Ceca. Genome Announc. 2016, 4, e00448-16. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, B.J.; Tai, A.Y.; Kotsanas, D.; Francis, M.J.; Roberts, S.A.; Ballard, S.A.; Junckerstorff, R.K.; Kormana, T.M. Clinical and microbiological characteristics of eggerthella lenta bacteremia. J. Clin. Microbiol. 2015, 53, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Bisanz, J.E.; Soto-Perez, P.; Noecker, C.; Aksenov, A.A.; Lam, K.N.; Kenney, G.E.; Bess, E.N.; Haiser, H.J.; Kyaw, T.S.; Yu, F.B.; et al. A genomic toolkit for the mechanistic dissection of intractable human gut bacteria. Cell Host Microbe 2020, 27, 1001. [Google Scholar] [CrossRef]
- La Reau, A.J.; Suen, G. The Ruminococci: Key symbionts of the gut ecosystem. J. Microbiol. 2018, 56, 199–208. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Zhang, Y.; Xiao, K.; Jiang, F.; Wang, H.; Tang, D.; Liu, D.; Liu, B.; Liu, Y.; He, X.; et al. The chicken gut metagenome and the modulatory effects of plant-derived benzylisoquinoline alkaloids. Microbiome 2018, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.D.; Hoyles, L.; Foster, G.; Falsen, E. Corynebacterium caspium sp. nov., from a Caspian seal (Phoca caspica). Int. J. Syst. Evol. Microbiol. 2004, 54, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Chapartegui-González, I.; Fernández-Martínez, M.; Rodríguez-Fernández, A.; Rocha, D.J.P.; Aguiar, E.R.G.R.; Pacheco, L.G.C.; Ramos-Vivas, J.; Calvo, J.; Martínez-Martínez, L.; Navas, J. Antimicrobial Susceptibility and Characterization of Resistance Mechanisms of Corynebacteriumurealyticum Clinical Isolates. Antibiotics 2020, 9, 404. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.M.; Freire, M.O.; Gabrilska, R.A.; Rumbaugh, K.P.; Lemon, K.P. Staphylococcus aureus Shifts toward Commensalism in Response to Corynebacterium Species. Front. Microbiol. 2016, 7, 1230. [Google Scholar] [CrossRef] [Green Version]
- La Reau, A.J.; Meier-Kolthoff, J.P.; Suen, G. Sequence-based analysis of the genus Ruminococcus resolves its phylogeny and reveals strong host association. Microb. Genomics 2016, 2, e000099. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Sadowsky, M.J. Understanding the mechanisms of faecal microbiota transplantation. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.; Guerrero-Araya, E.; Cortés-Tapia, C.; Plaza-Garrido, Á.; Lawley, T.D.; Paredes-Sabja, D. Comprehensive genome analyses of Sellimonas intestinalis, a potential biomarker of homeostasis gut recovery. bioRxiv 2020, 6, 12. [Google Scholar] [CrossRef]
- De Vos, W.M. Systems solutions by lactic acid bacteria: From paradigms to practice. Microb. Cell Fact. 2011, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Haberecht, S.; Bajagai, Y.S.; Moore, R.J.; Van, T.T.H.; Stanley, D. Poultry feeds carry diverse microbial communities that influence chicken intestinal microbiota colonisation and maturation. AMB Express 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Baqi, M.; Mazzulli, T. Oligella infections: Case report and review of the literature. Can. J. Infect. Dis. 1996, 7, 377. [Google Scholar] [CrossRef] [Green Version]
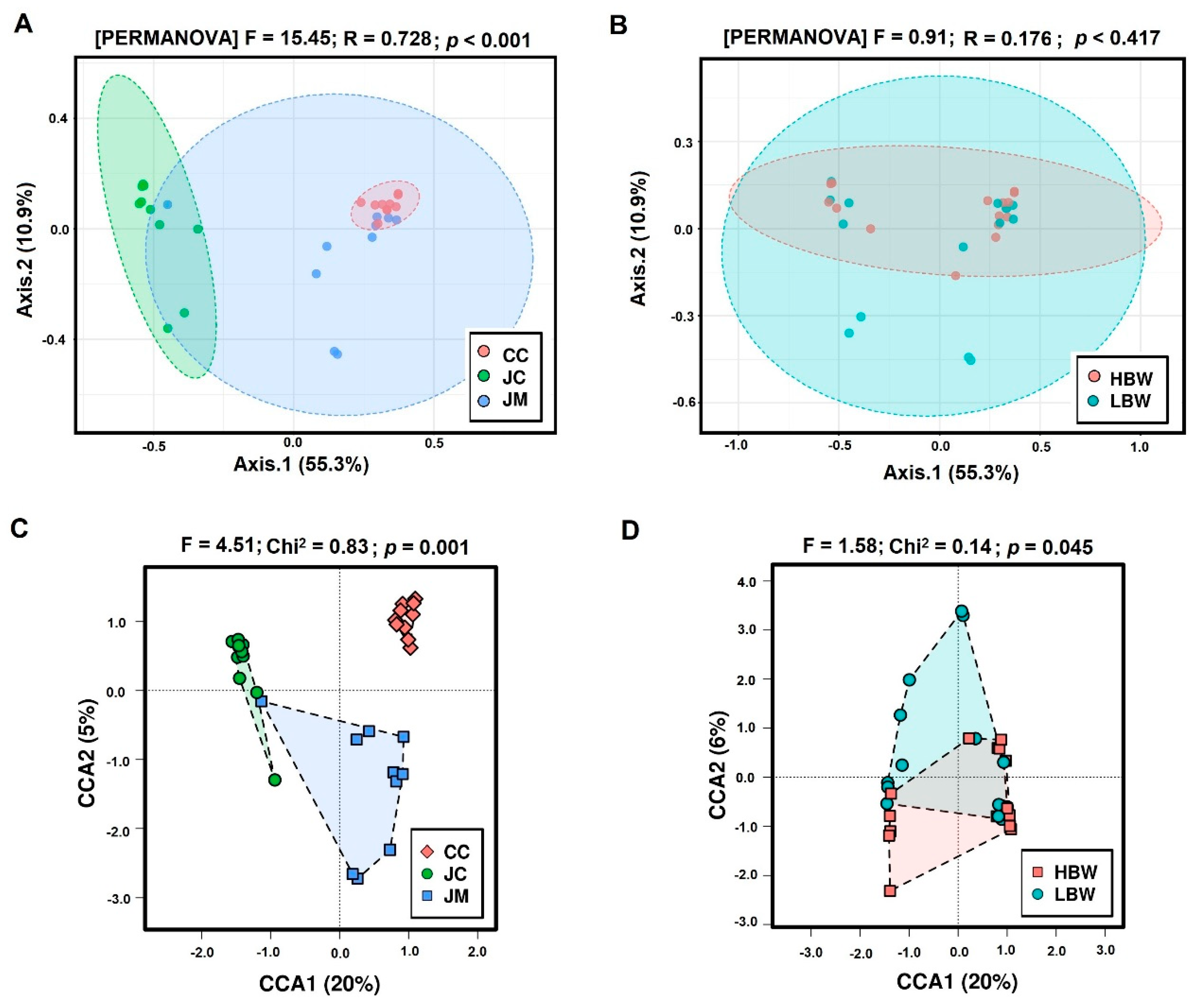

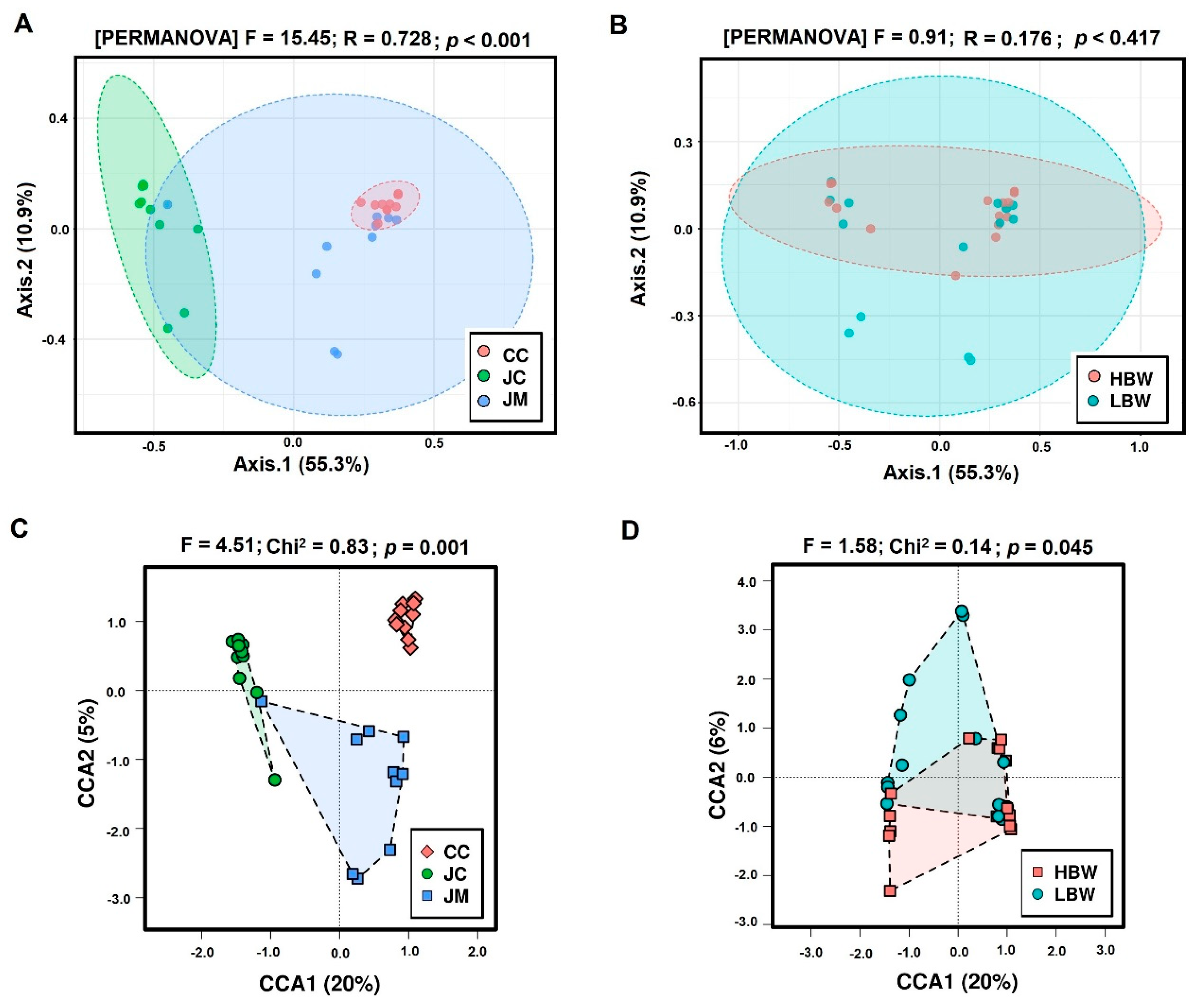{kind=link}
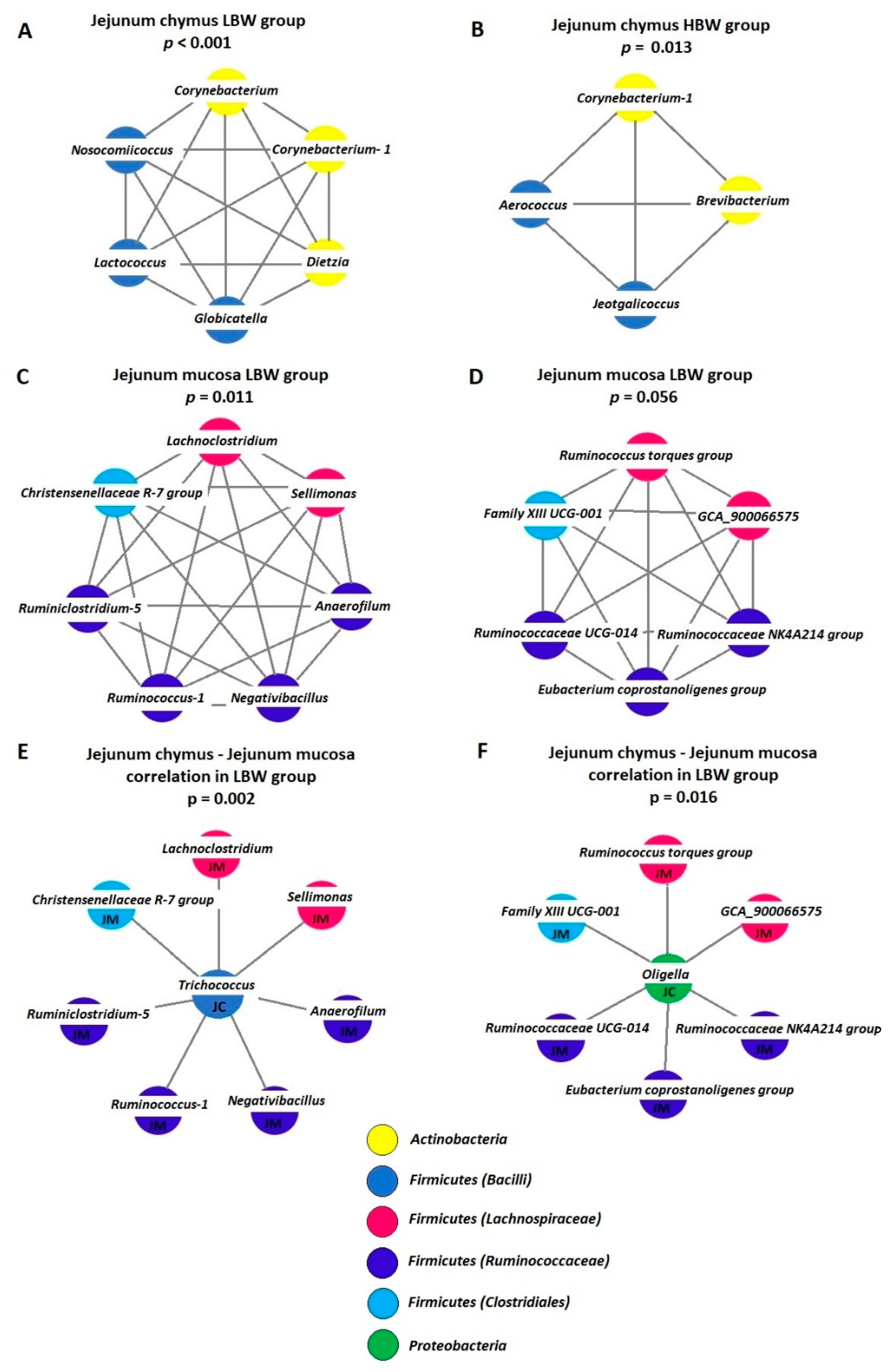{kind=link}
| SP | BW | Diversity Indices (Mean) | ||||
|---|---|---|---|---|---|---|
| OTU | Chao1 | Shannon | Simpson | PD | ||
| JC | LBW | 105 | 106 | 2.86 | 0.71 | 38.0 |
| HBW | 122 | 122 | 3.17 | 0.72 | 41.8 | |
| JM | LBW | 345 | 346 | 5.11 | 0.82 | 101.6 |
| HBW | 360 | 361 | 6.45 | 0.97 | 109.2 | |
| CC | LBW | 362 | 363 | 6.10 | 0.963 | 97.9 |
| HBW | 350 | 351 | 5.98 | 0.955 | 97.5 | |
| JC | 111 b | 111 b | 2.08 b | 0.71 b | 39.9 b | |
| JM | 345 a | 346 a | 3.97 a | 0.89 a | 105.4 a | |
| CC | 355 a | 357 a | 4.18 a | 0.96 a | 97.7 a | |
| LBW | 269 | 270 | 3.24 | 0.83 | 79.2 | |
| HBW | 272 | 272 | 3.59 | 0.88 | 82.9 | |
| Pooled SEM | 23.67 | 23.74 | 0.31 | 0.03 | 6.25 | |
| p-Values | SP | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 |
| BW | 0.779 | 0.780 | 0.162 | 0.256 | 0.589 | |
| SP × BW | 0.860 | 0.863 | 0.247 | 0.311 | 0.891 | |
| Body Weight | Sampling Place | Mean (BW) | FDR p-Values | |||||
|---|---|---|---|---|---|---|---|---|
| Jejunal Chymus | Jejunal Mucosa | Caecum Chymus | SP | BW | SP × BW | |||
| Actinobacteria | LBW | 2.54 | 1.55 | 0.38 | 1.49 | 0.188 | 0.898 | 0.681 |
| HBW | 4.40 | 0.46 | 0.17 | 1.67 | ||||
| Mean (SP) | 3.47 | 1.00 | 0.28 | |||||
| Bacteroidetes | LBW | 0.38 | 20.83 B | 45.25 | 22.15 B | <0.001 | 0.029 | 0.109 |
| HBW | 1.18 | 39.99 A | 51.61 | 30.93 A | ||||
| Mean (SP) | 0.78 c | 30.41 b | 48.43 a | |||||
| Cyanobacteria | LBW | 4.82 | 0.47 | 0.05 | 1.78 | 0.163 | 0.990 | 0.878 |
| HBW | 3.76 | 1.47 | 0.06 | 1.76 | ||||
| Mean (SP) | 4.29 | 0.97 | 0.06 | |||||
| Deinococcus Thermus | LBW | 0.04 | 0.02 | 0.00 | 0.02 | 0.406 | 0.532 | 0.843 |
| HBW | 0.02 | 0.00 | 0.00 | 0.01 | ||||
| Mean (SP) | 0.03 | 0.01 | 0.00 | |||||
| Epsilonbacteraeota | LBW | 0.00 | 0.00 | 0.00 | 0.00 | 0.567 | 0.525 | 0.580 |
| HBW | 0.01 | 0.11 | 0.00 | 0.04 | ||||
| Mean (SP) | 0.01 | 0.06 | 0.00 | |||||
| Firmicutes | LBW | 88.16 | 74.76 A | 51.77 | 71.57 A | <0.001 | 0.053 T | 0.532 |
| HBW | 76.67 | 54.78 B | 46.92 | 59.46 B | ||||
| Mean (SP) | 82.42 a | 64.77 b | 49.35 c | |||||
| Patescibacteria | LBW | 0.25 | 0.05 | 0.00 | 0.1 | 0.530 | 0.530 | 0.559 |
| HBW | 4.03 | 0.12 | 0.00 | 1.38 | ||||
| Mean (SP) | 2.14 | 0.09 | 0.00 | |||||
| Proteobacteria | LBW | 3.78 | 2.25 | 2.39 | 2.81 | 0.045 | 0.399 | 0.170 |
| HBW | 9.88 | 2.72 | 0.98 | 4.53 | ||||
| Mean (SP) | 6.83 a | 2.48 b | 1.68 b | |||||
| Tenericutes | LBW | 0.01 | 0.08 B | 0.15 B | 0.08 B | 0.056 T | 0.068 T | 0.406 |
| HBW | 0.05 | 0.34 A | 0.25 A | 0.21 A | ||||
| Mean (SP) | 0.03 b | 0.21 a | 0.20 a | |||||
| B/F Ratio | LBW | 0.00 | 0.32 B | 0.89 | 0.40 B | <0.001 | 0.029 | 0.188 |
| HBW | 0.02 | 0.76 A | 1.13 | 0.64 A | ||||
| Mean (SP) | 0.01 c | 0.54 b | 1.01 a | |||||
| Genus | Body Weight | Sampling Place | Mean (BW) | FDR p-Values | ||||
|---|---|---|---|---|---|---|---|---|
| Jejunal Chymus | Jejunal Mucosa | Caecum Chymus | SP | BW | SP × BW | |||
| Alistipes | LBW | 0.00 | 0.64 | 0.82 B | 0.48 B | 0.451 | ||
| HBW | 0.03 | 1.71 | 2.48 A | 1.41 A | 0.081 T | |||
| Mean (SP) | 0.01 b | 1.17 a | 1.65 a | 0.017 | ||||
| Bacteroides | LBW | 0.11 | 19.06 B | 42.39 | 20.52 B | 0.280 | ||
| HBW | 0.76 | 34.28 A | 46.27 | 27.10 A | 0.098 T | |||
| Mean (SP) | 0.43 c | 26.67 b | 44.33 a | 0.000 | ||||
| Enterococcus | LBW | 0.02 B | 0.03 | 0.00 | 0.02 B | 0.001 | ||
| HBW | 0.22 A | 0.02 | 0.00 | 0.08 A | 0.012 | |||
| Mean (SP) | 0.123 a | 0.025 b | 0.002 b | 0.000 | ||||
| Ruminococcaceae UCG-010 | LBW | 0.00 | 0.09 B | 0.19 | 0.09 B | 0.472 | ||
| HBW | 0.00 | 0.24 A | 0.29 | 0.18 A | 0.083 T | |||
| Mean (SP) | 0.00 b | 0.17 a | 0.24 a | 0.000 | ||||
| Ruminococcaceae UCG-013 | LBW | 0.01 | 0.09 | 0.39 A | 0.17 A | 0.026 | ||
| HBW | 0.01 | 0.06 | 0.18 B | 0.08 B | 0.020 | |||
| Mean (SP) | 0.010 b | 0.078 b | 0.285 a | 0.000 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farkas, V.; Csitári, G.; Menyhárt, L.; Such, N.; Pál, L.; Husvéth, F.; Rawash, M.A.; Mezőlaki, Á.; Dublecz, K. Microbiota Composition of Mucosa and Interactions between the Microbes of the Different Gut Segments Could Be a Factor to Modulate the Growth Rate of Broiler Chickens. Animals 2022, 12, 1296. https://doi.org/10.3390/ani12101296
Farkas V, Csitári G, Menyhárt L, Such N, Pál L, Husvéth F, Rawash MA, Mezőlaki Á, Dublecz K. Microbiota Composition of Mucosa and Interactions between the Microbes of the Different Gut Segments Could Be a Factor to Modulate the Growth Rate of Broiler Chickens. Animals. 2022; 12(10):1296. https://doi.org/10.3390/ani12101296
Chicago/Turabian StyleFarkas, Valéria, Gábor Csitári, László Menyhárt, Nikoletta Such, László Pál, Ferenc Husvéth, Mohamed Ali Rawash, Ákos Mezőlaki, and Károly Dublecz. 2022. "Microbiota Composition of Mucosa and Interactions between the Microbes of the Different Gut Segments Could Be a Factor to Modulate the Growth Rate of Broiler Chickens" Animals 12, no. 10: 1296. https://doi.org/10.3390/ani12101296
APA StyleFarkas, V., Csitári, G., Menyhárt, L., Such, N., Pál, L., Husvéth, F., Rawash, M. A., Mezőlaki, Á., & Dublecz, K. (2022). Microbiota Composition of Mucosa and Interactions between the Microbes of the Different Gut Segments Could Be a Factor to Modulate the Growth Rate of Broiler Chickens. Animals, 12(10), 1296. https://doi.org/10.3390/ani12101296