
De Novo Transcriptome Assembly, Functional Annotation and SSR Marker Discovery of Qinling Takin (Budorcas taxicolor bedfordi)


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation for Transcriptome Sequencing
2.2. De Novo Assembly
2.3. Functional Annotation
2.4. Identification of SSR
2.5. Phylogenetic Analysis
3. Results and Discussion
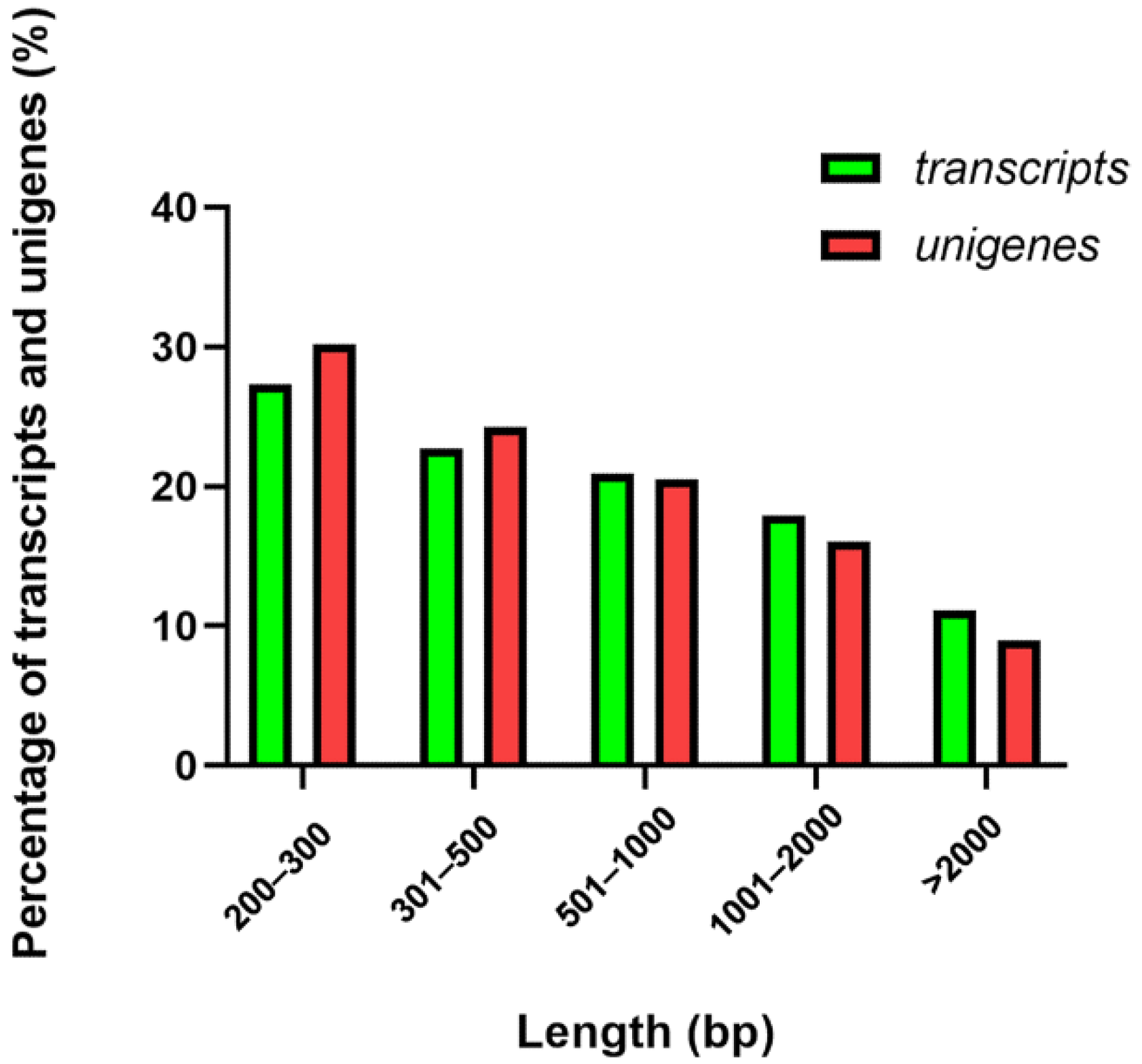
3.1. Sequencing and De Novo Assembly of Takin Transcriptome
3.2. Functional Annotation
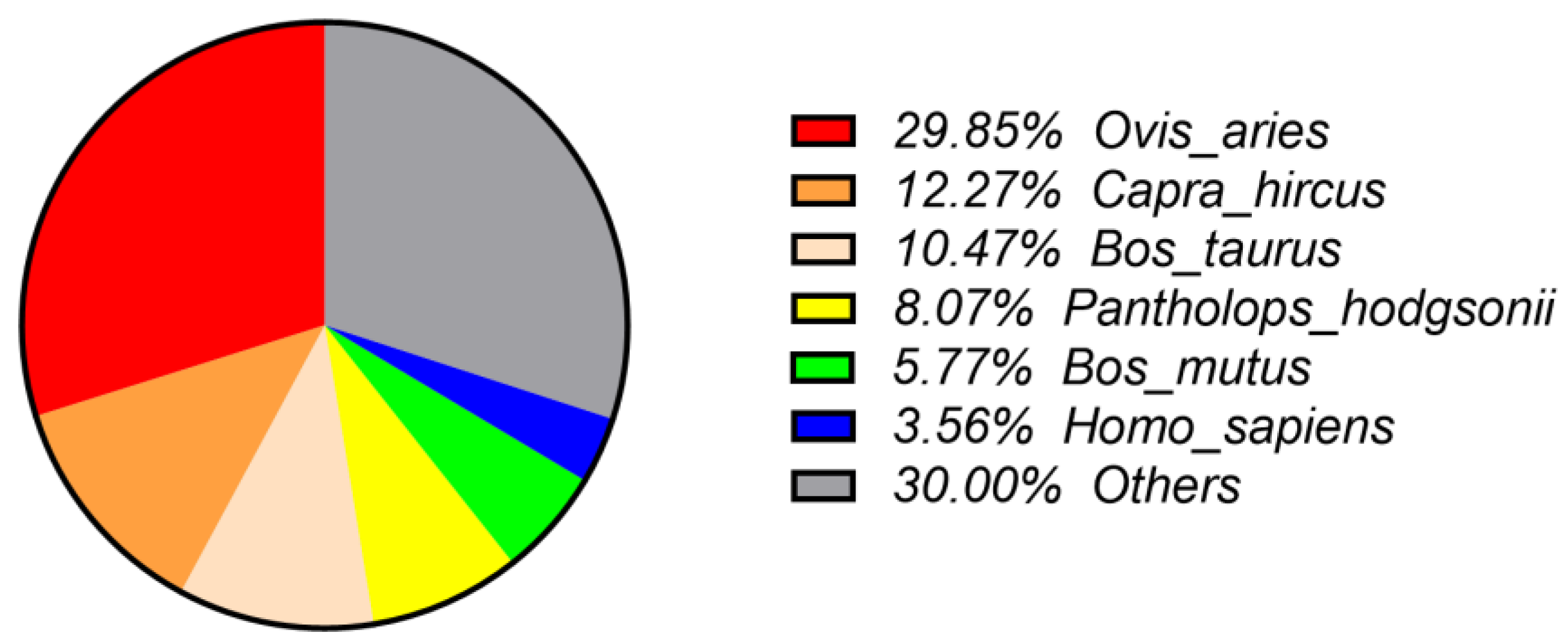
3.2.1. NR Annotation
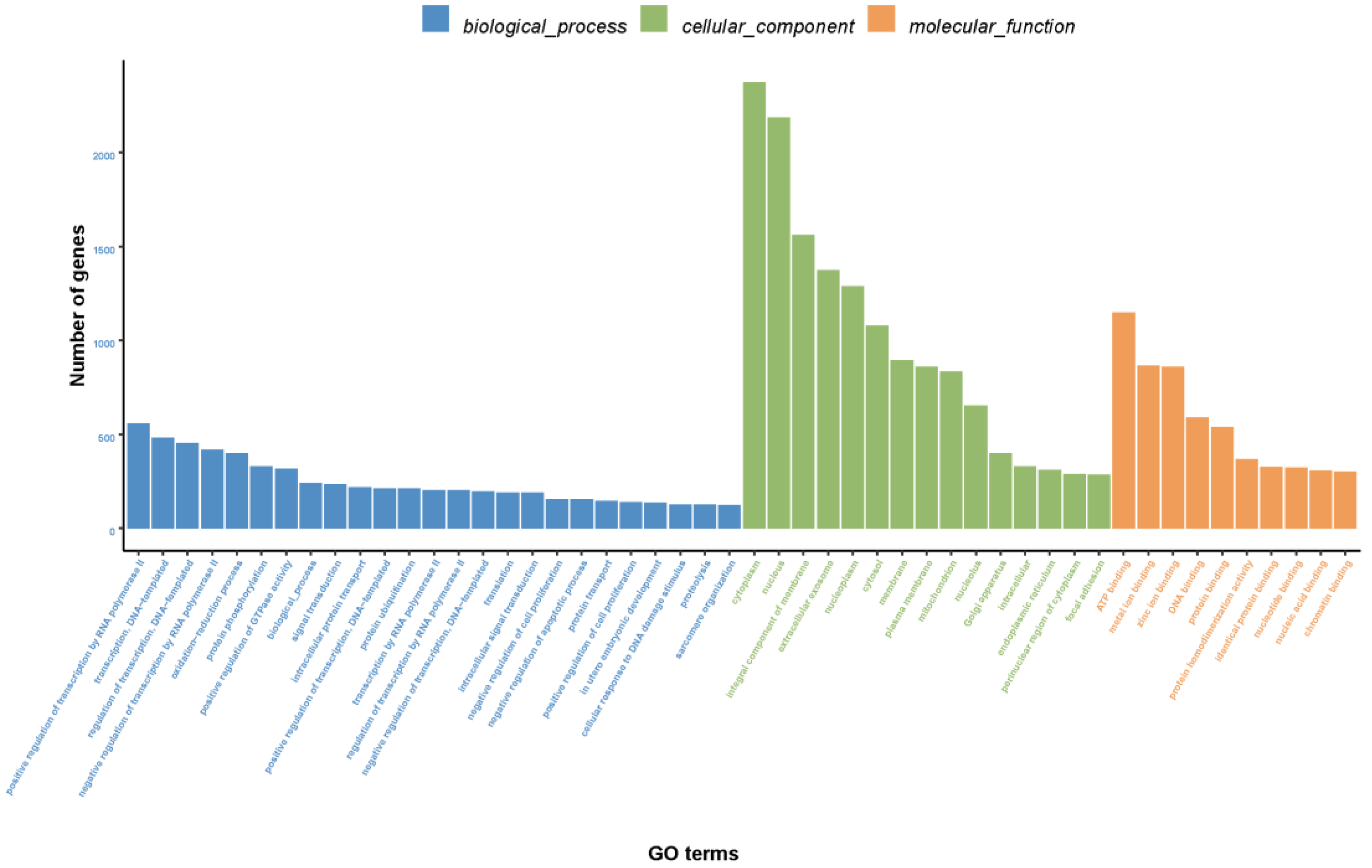
3.2.2. GO Annotation
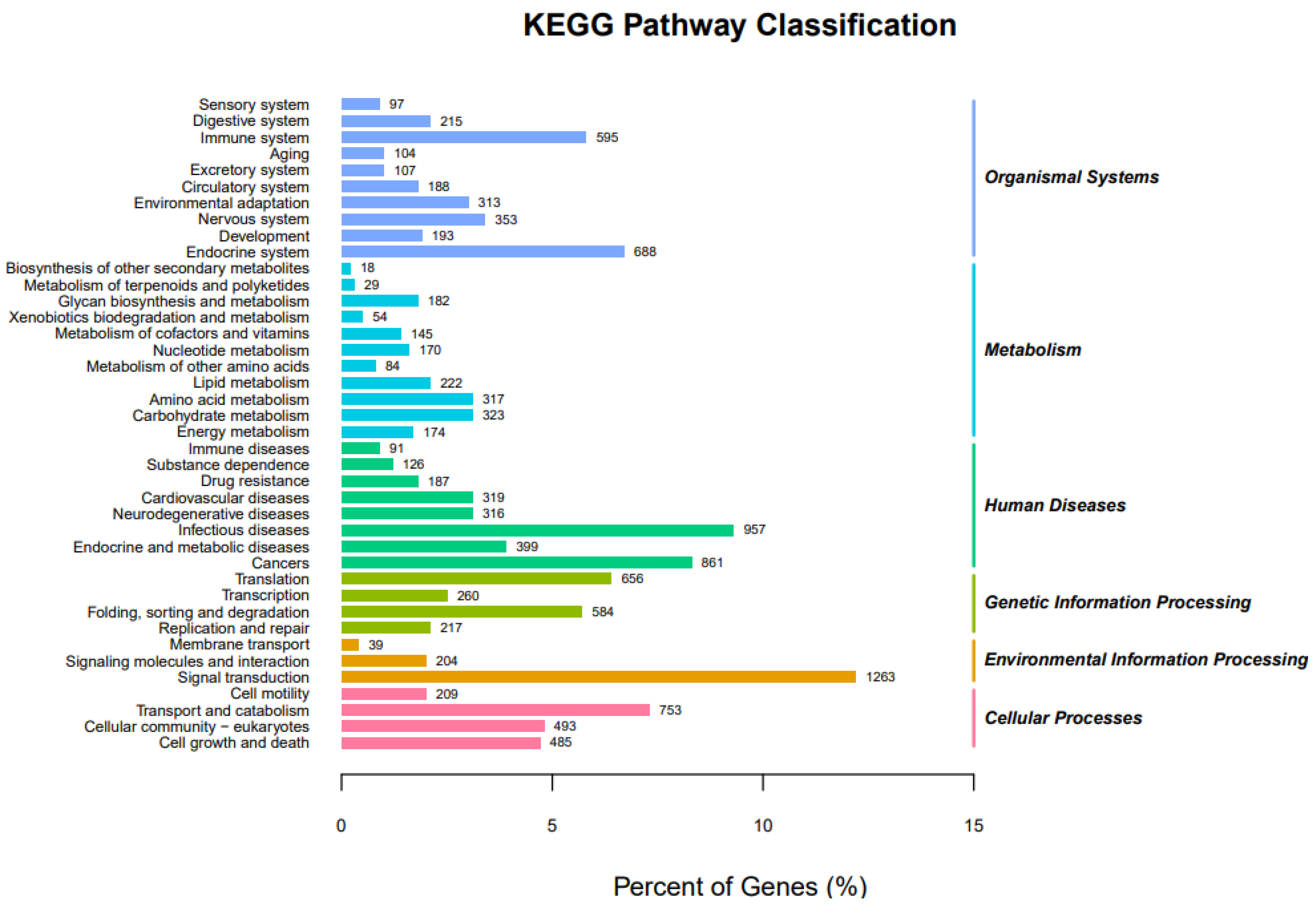
3.2.3. KEGG Pathway
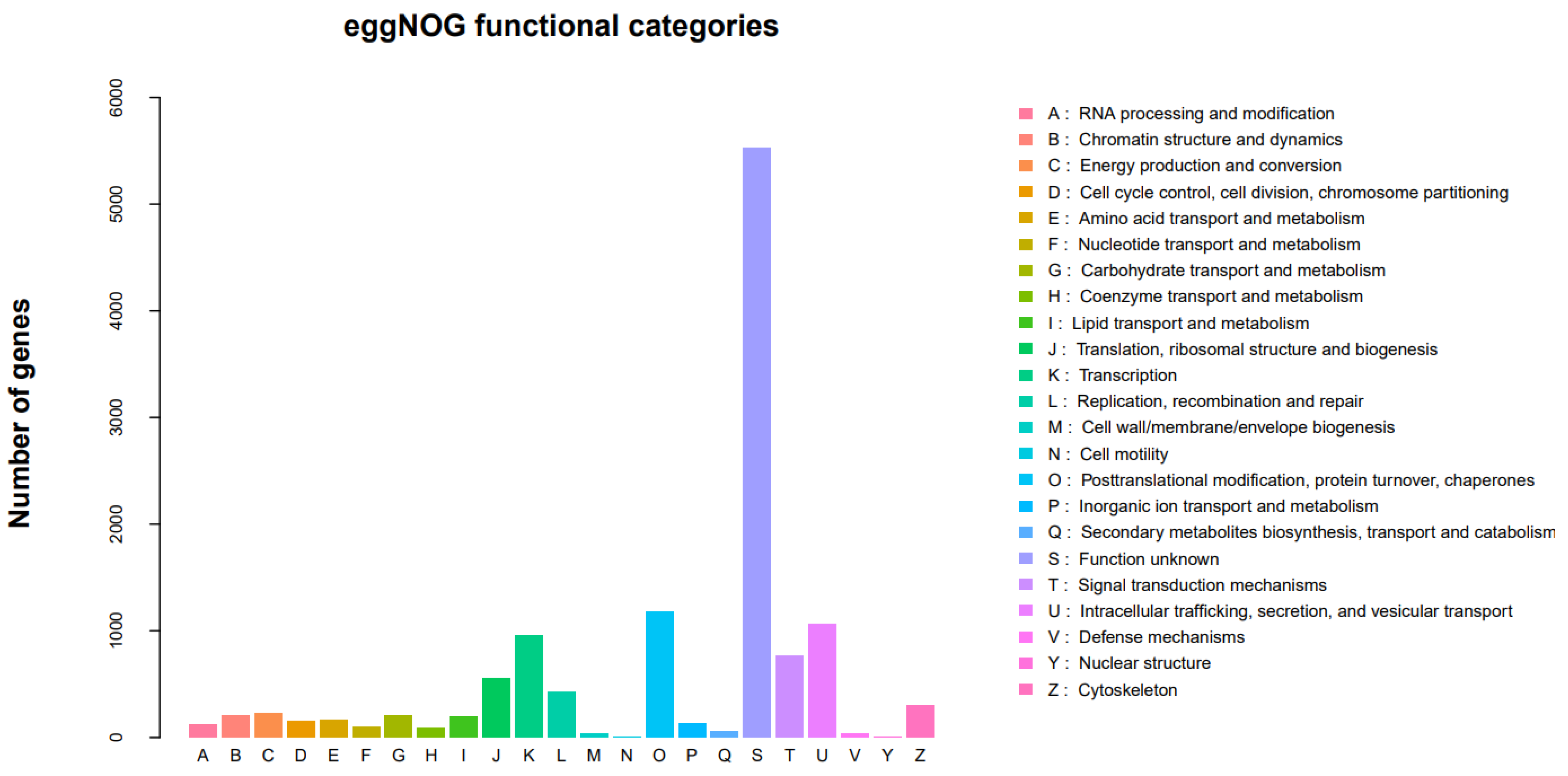
3.2.4. EggNOG Classification
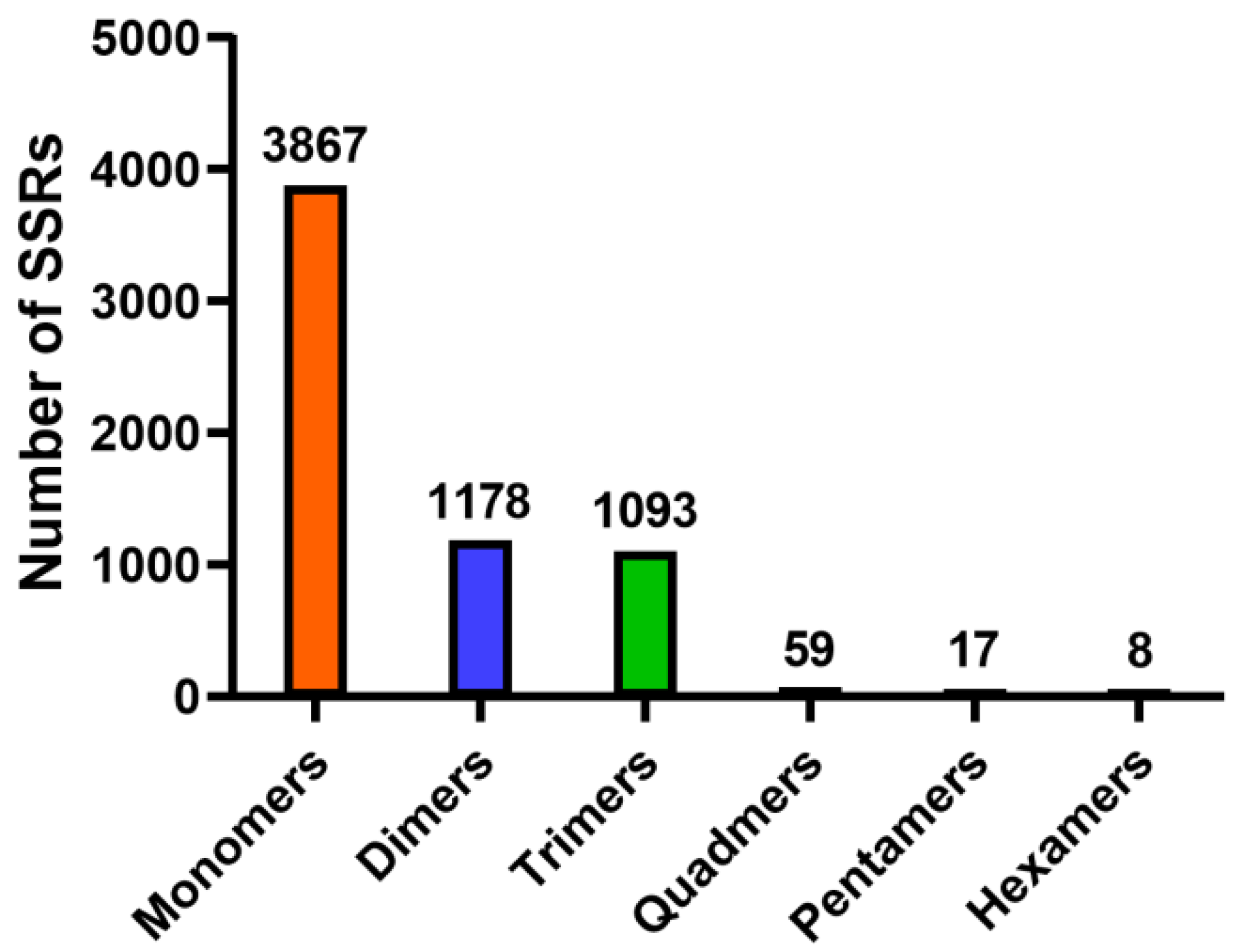
3.3. Identification of SSR Markers
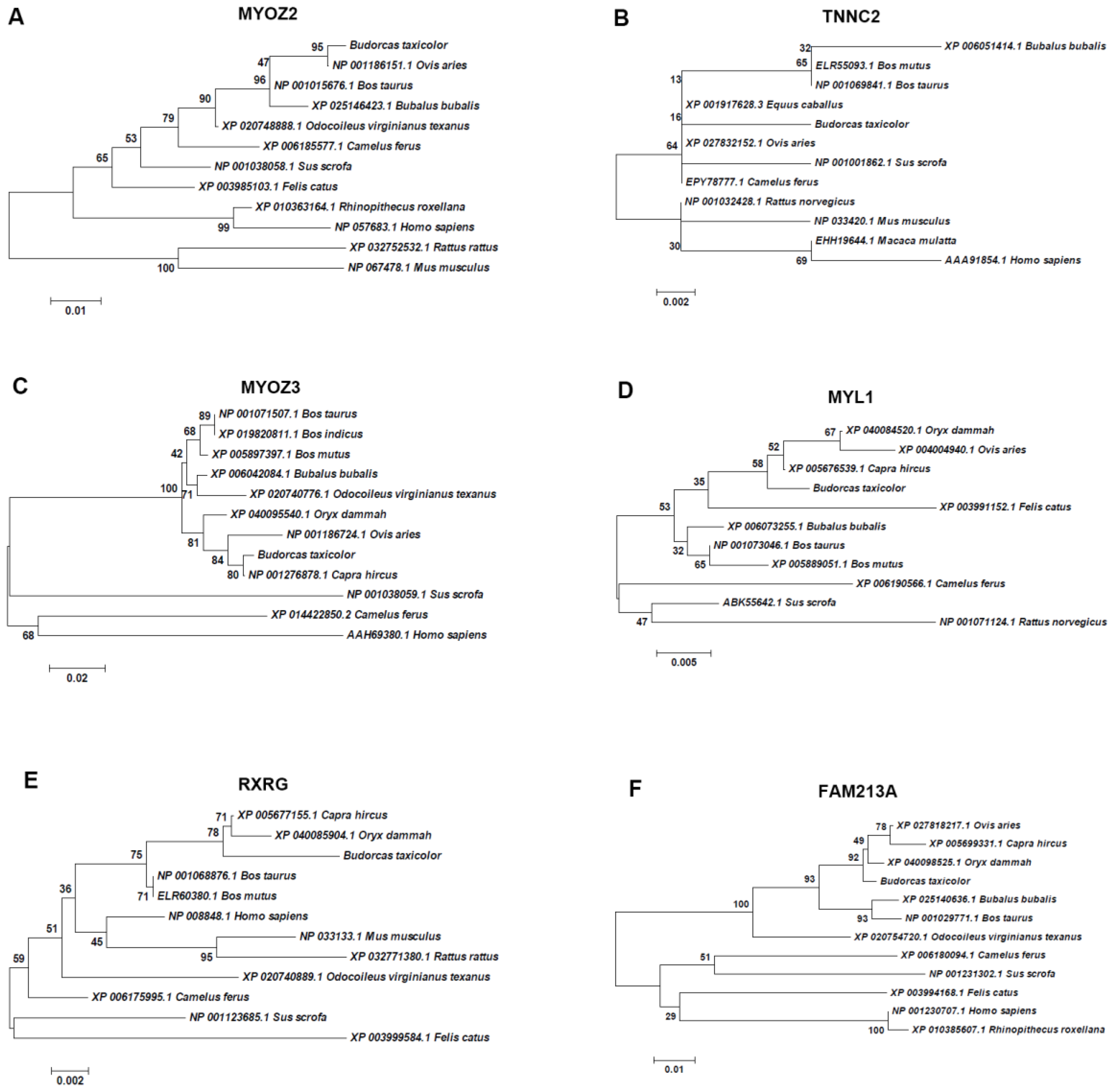
3.4. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, J. Study of system and distribution of Chinese Takin. Zool. Res. 1986, 7, 167–175. [Google Scholar]
- Li, M.; Meng, S.; Wei, F.; Wang, J.; Yong, Y. Genetic diversity and population genetic structure of Takin (Budorcas taxicolor). Acta Theriol. Sin. 2003, 23, 10–16. [Google Scholar]
- Zeng, Z.; Song, Y. Ecology and protection of Qinling Takin. Bull. Biol. 2008, 43, 1–4, 63. [Google Scholar]
- Zeng, Z.; Song, Y. Golden Takin (Budorcas taxicolor bedfordi). Chin. J. Zool. 2002, 37, 5. [Google Scholar]
- Gentry, A.W. The subfamilies and tribes of the family Bovidae. Mammal. Rev. 1992, 22, 1–32. [Google Scholar] [CrossRef]
- Pasitschniak-Arts, M.; Flood, P.F.; Schmutz, S.M.; Seidel, B. A comparison of G-band patterns of the muskox and takin and their evolutionary relationship to sheep. J. Hered. 1994, 85, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Groves, P.; Shields, G.F. Cytochrome B sequences suggest convergent evolution of the Asian takin and Arctic muskox. Mol. Phylogenetics Evol. 1997, 8, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Wang, J.; Liu, P.; Su, B.; Zhang, Y. Analysis of cytochrome b gene of takin and its phylogeny. J. Northwest. Univ. (Nat. Sci. Ed. ) 2001, 31, 347–350. [Google Scholar]
- Ren, Y.; Feng, H.; Feng, C.; Meng, S. Phylogenic analysis of Takin by mtDNA. Genom. Appl. Biol. 2012, 31, 481–484. [Google Scholar]
- Feng, H.; Feng, C.; Huang, Y.; Wang, L. Analysis on complete mitochondrial genome sequence of golden Takin (Budorcas taxicolor). Genom. Appl. Biol. 2016, 35, 2299–2305. [Google Scholar]
- Kumar, A.; Gautam, K.B.; Singh, B.; Yadav, P.; Gopi, G.V.; Gupta, S.K. Sequencing and characterization of the complete mitochondrial genome of Mishmi takin (Budorcas taxicolor taxicolor) and comparison with the other Caprinae species. Int. J. Biol. Macromol. 2019, 137, 87–94. [Google Scholar] [CrossRef]
- Zhou, M.; Yu, J.; Li, B.; Ouyang, B.; Yang, J. The complete mitochondrial genome of Budorcas taxicolor tibetana (Artiodactyla: Bovidae) and comparison with other Caprinae species: Insight into the phylogeny of the genus Budorcas. Int. J. Biol. Macromol. 2019, 121, 223–232. [Google Scholar] [CrossRef]
- Feldmesser, E.; Rosenwasser, S.; Vardi, A.; Ben-Dor, S. Improving transcriptome construction in non-model organisms: Integrating manual and automated gene definition in Emiliania huxleyi. BMC Genom. 2014, 15, 148. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Zhu, L.; Nie, H.; Yin, M.; Liu, G.; Yan, X. De novo assembly, gene annotation, and marker development using Illumina paired-end transcriptome sequencing in the Crassadoma gigantea. Gene 2018, 658, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, H.; Duan, X.; Hu, J.; Li, J.; Zhao, L.; Ma, Y. Characterizing the Leaf Transcriptome of Chrysanthemum rhombifolium (Ling et C. Shih), a Drought Resistant, Endemic Plant From China. Front. Genet. 2021, 12, 625985. [Google Scholar] [CrossRef]
- Ceschin, D.G.; Pires, N.S.; Mardirosian, M.N.; Lascano, C.I.; Venturino, A. The Rhinella arenarum transcriptome: De novo assembly, annotation and gene prediction. Sci. Rep. 2020, 10, 1053. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, X.; Qiu, Z.; Huang, Y. De Novo Assembly and Characterization of the Xenocatantops brachycerus Transcriptome. Int. J. Mol. Sci. 2018, 19, 520. [Google Scholar] [CrossRef]
- Zhu, C.; Pan, Z.; Wang, H.; Chang, G.; Wu, N.; Ding, H. De novo assembly, characterization and annotation for the transcriptome of Sarcocheilichthys sinensis. PLoS ONE 2017, 12, e0171966. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Pang, C.; Lu, X.; Zhu, P.; Duan, A.; Tan, Z.; Huang, J.; Li, H.; Chen, M.; Liang, X. De Novo Transcriptome Assembly of the Chinese Swamp Buffalo by RNA Sequencing and SSR Marker Discovery. PLoS ONE 2016, 11, e0147132. [Google Scholar] [CrossRef] [PubMed]
- Song, X.J.; Jiang, H.Y.; Qi, Z.; Shen, X.; Xue, M.; Hu, J.G.; Liu, H.M.; Zhou, X.H.; Tu, J.; Qi, K.Z. APEC infection affects cytokine-cytokine receptor interaction and cell cycle pathways in chicken trachea. Res. Vet. Sci. 2020, 130, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhang, J.; Liang, X.; Zhang, X.; Wang, T.; Yin, S. Integrated Analysis of Transcriptomic, miRNA and Proteomic Changes of a Novel Hybrid Yellow Catfish Uncovers Key Roles for miRNAs in Heterosis. Mol. Cell. Proteom. 2019, 18, 1437–1453. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Li, A.; Chen, Y.; Zhao, X.; Niu, Y.; Cong, P.; Zhang, Z.; Chen, W.; Jiang, W.; Mo, D. Characterization and transcriptional regulation analysis of the porcine TNFAIP8L2 gene. Mol. Genet. Genom. 2010, 284, 185–195. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Hou, L.; Ji, Z.; Wang, J. De novo assembly and characterization of the skeletal muscle transcriptome of sheep using Illumina paired-end sequencing. Biotechnol. Lett. 2015, 37, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Dou, Z.; Gu, Y.; Zhang, Q.; Lu, R.; Zhu, X. Cloning and analysis of highly repetitive sequence fragments from Takin (Budorcas taxicolor). Acta Biol. Exp. Sin. 2002, 35, 36–41. [Google Scholar]
- Li, X.; Liu, X.; Wei, J.; Li, Y.; Tigabu, M.; Zhao, X. Development and Transferability of EST-SSR Markers for Pinus koraiensis from Cold-Stressed Transcriptome through Illumina Sequencing. Genes 2020, 11, 500. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, B.A.; Agapito, A.M.; Novoa-Munoz, F.; Vianna, J.; Johnson, W.E.; Marin, J.C. Utility of genetic variation in coat color genes to distinguish wild, domestic and hybrid South American camelids for forensic and judicial applications. Forensic Sci. Int. Genet. 2020, 45, 102226. [Google Scholar] [CrossRef]
- Tahoor, A.; Khan, J.A.; Mahfooz, S. A comparative survey of microsatellites among wild and domestic cat provides valuable resources for marker development. Mol. Biol. Rep. 2019, 46, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; Dettleff, P.; Valenzuela, C.; Estrada, J.M.; Valdes, J.A.; Meneses, C.; Molina, A. Evaluating the genetic structure of wild and commercial red cusk-eel (Genypterus chilensis) populations through the development of novel microsatellite markers from a reference transcriptome. Mol. Biol. Rep. 2019, 46, 5875–5882. [Google Scholar] [CrossRef] [PubMed]
- Moura, R.F.; Dawson, D.A.; Nogueira, D.M. The use of microsatellite markers in Neotropical studies of wild birds: A literature review. Acad. Bras. Cienc. 2017, 89, 145–154. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Number/Length |
|---|---|
| Raw reads | 56,745,076 |
| Raw bases (Gb) | 8.51 |
| Clean reads | 55,417,300 |
| Clean bases (Gb) | 7.76 |
| Q20 percentage (%) | 98.39 |
| Q30 percentage (%) | 94.43 |
| GC percentage (%) | 53.13 |
| Transcripts | 25,677 |
| N50 length (bp) | 1596 |
| Mean length (bp) | 925 |
| Unigenes | 21,648 |
| N50 length (bp) | 1388 |
| Mean length (bp) | 817 |
| Database | Number of Unigenes | Ratio (%) |
|---|---|---|
| All | 21,648 | 100.00 |
| NR | 13,214 | 61.04 |
| GO | 12,071 | 55.76 |
| KEGG | 10,329 | 47.71 |
| EggNOG | 12,157 | 56.16 |
| Swissprot | 11,606 | 53.61 |
| Pfam | 8890 | 41.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, J.; Guo, R.; Li, Y.; Zhang, Y.; Jia, K.; Lei, Y.; Zan, L.; Li, A. De Novo Transcriptome Assembly, Functional Annotation and SSR Marker Discovery of Qinling Takin (Budorcas taxicolor bedfordi). Animals 2021, 11, 2366. https://doi.org/10.3390/ani11082366
Qiu J, Guo R, Li Y, Zhang Y, Jia K, Lei Y, Zan L, Li A. De Novo Transcriptome Assembly, Functional Annotation and SSR Marker Discovery of Qinling Takin (Budorcas taxicolor bedfordi). Animals. 2021; 11(8):2366. https://doi.org/10.3390/ani11082366
Chicago/Turabian StyleQiu, Ju, Rui Guo, Yidan Li, Yuyao Zhang, Kangsheng Jia, Yinghu Lei, Linsen Zan, and Anning Li. 2021. "De Novo Transcriptome Assembly, Functional Annotation and SSR Marker Discovery of Qinling Takin (Budorcas taxicolor bedfordi)" Animals 11, no. 8: 2366. https://doi.org/10.3390/ani11082366
APA StyleQiu, J., Guo, R., Li, Y., Zhang, Y., Jia, K., Lei, Y., Zan, L., & Li, A. (2021). De Novo Transcriptome Assembly, Functional Annotation and SSR Marker Discovery of Qinling Takin (Budorcas taxicolor bedfordi). Animals, 11(8), 2366. https://doi.org/10.3390/ani11082366