
Seasonal Variation in the Faecal Microbiota of Mature Adult Horses Maintained on Pasture in New Zealand

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Experimental Design
Animal Details and Management
2.3. Sample Collection
2.3.1. Faecal Sample Collection
2.3.2. Feed Sample Collection
2.3.3. Laboratory Analysis DNA Extraction, Amplicon Library Construction, and Sequencing
2.3.4. Analysis of Nutrient Composition
2.4. Data Recording and Analysis
2.4.1. Bioinformatics Analysis
2.4.2. Statistical Analyses
3. Results
3.1. Health and Condition of the Horses
3.2. Forage and Climate Monitoring
3.3. Comparison of the Faecal Microbiota (Bacterial Communities)
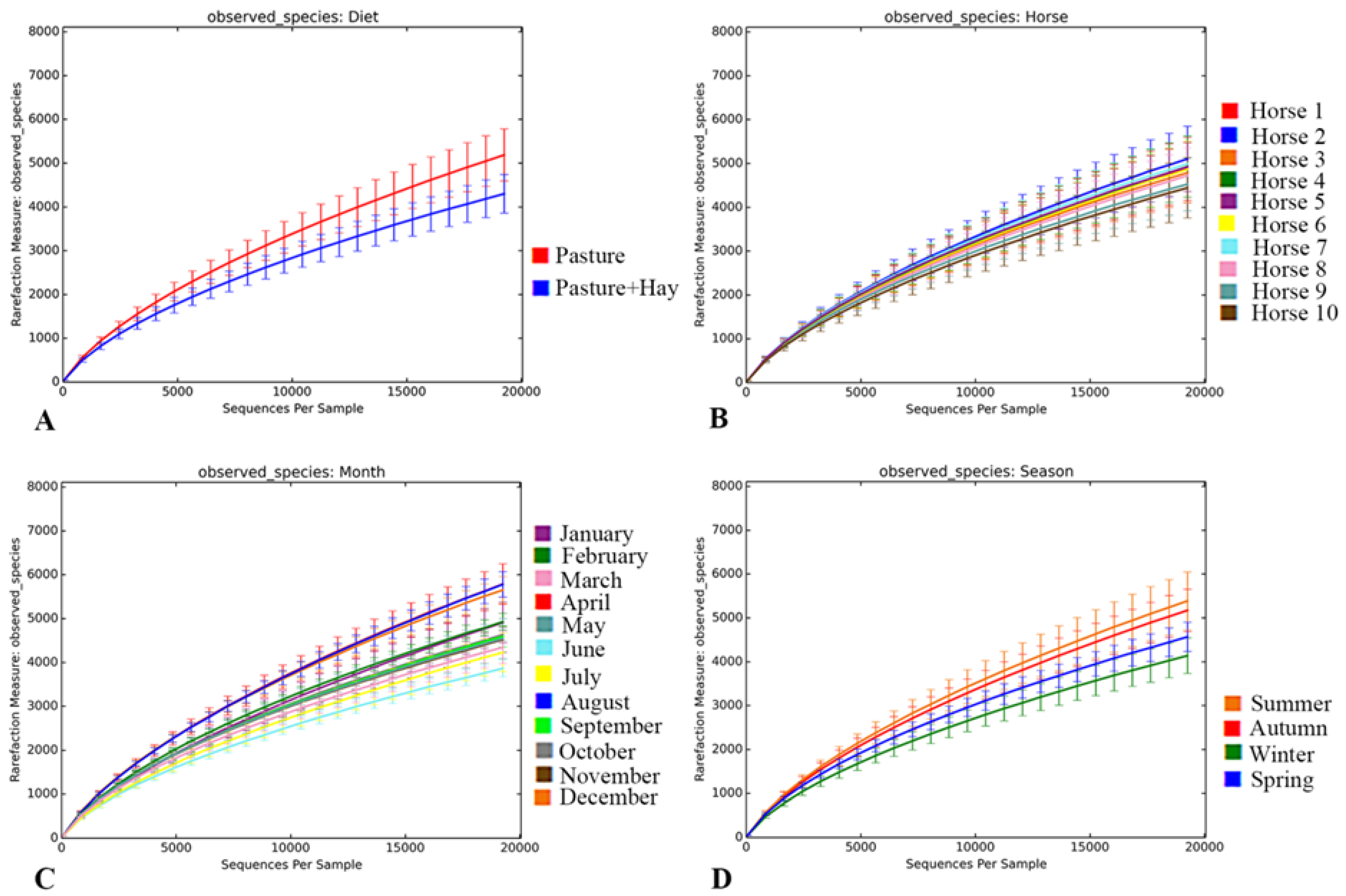
3.3.1. Metrics of Sequencing Data and Rarefaction Analysis
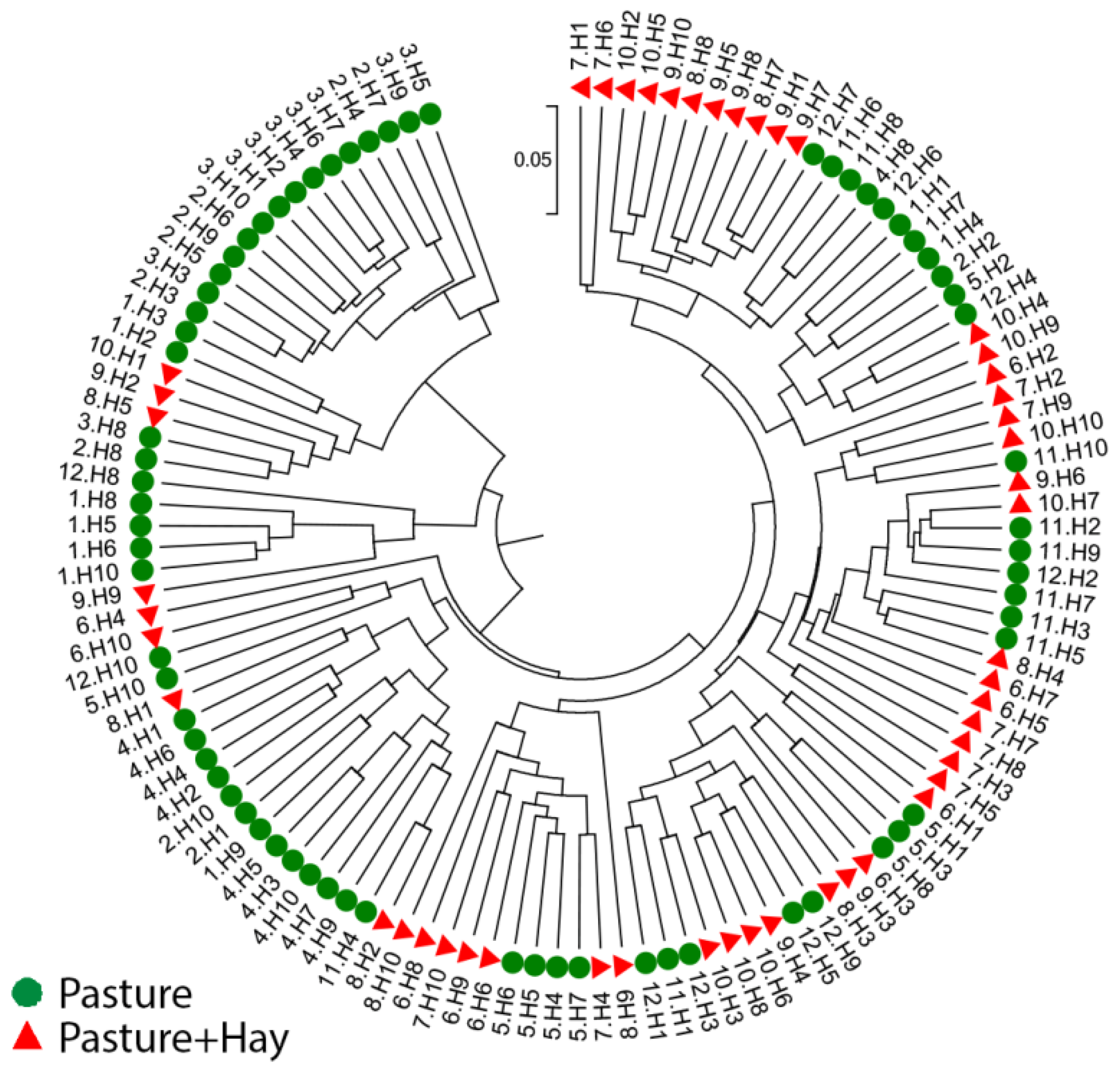
3.3.2. Diet-Specific Differences in Diversity of the Faecal Bacterial Community
3.3.3. Inter- and Intra-Horse Variation
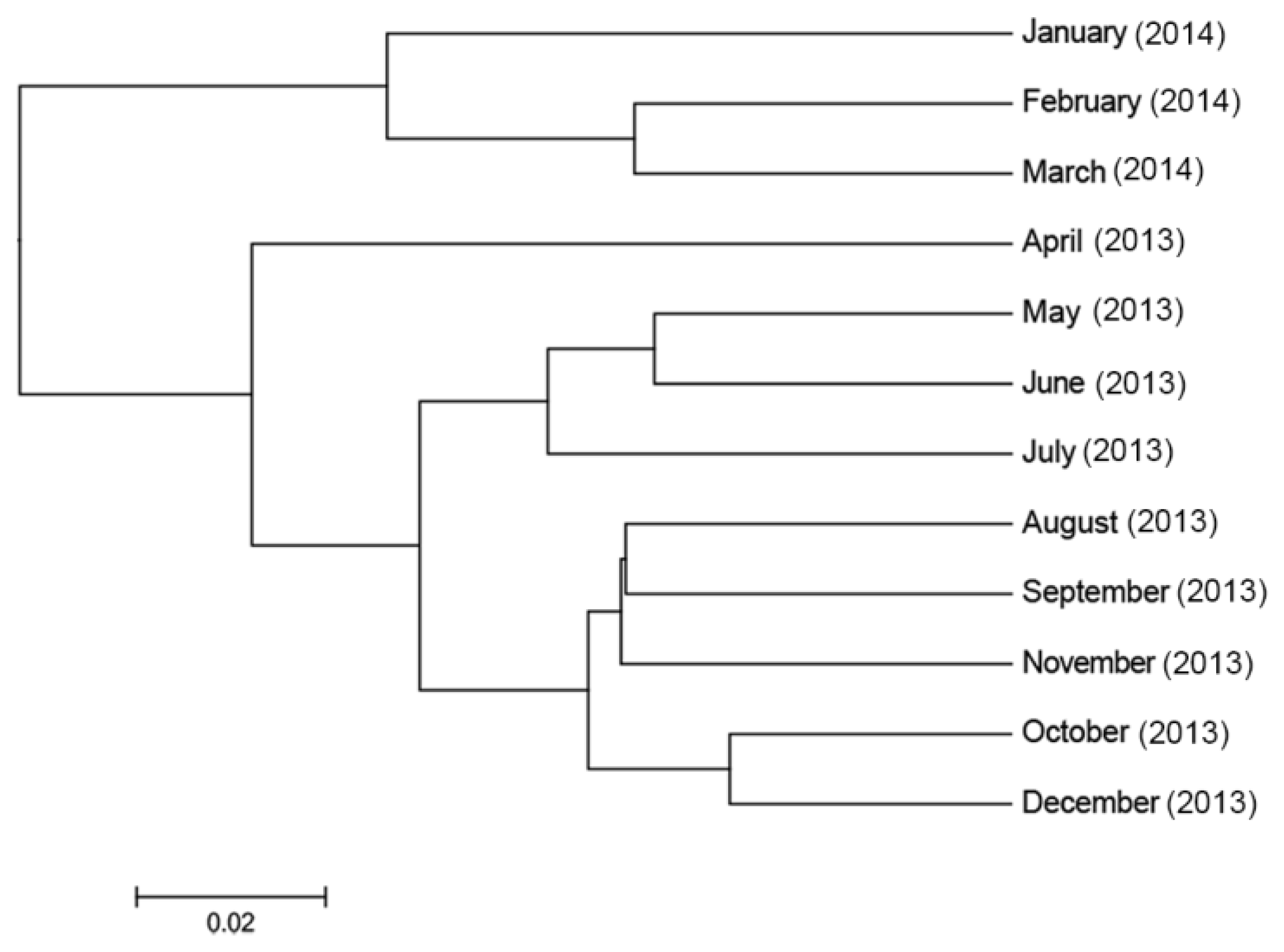
3.3.4. Temporal Effects on the Diversity of the Faecal Bacterial Community
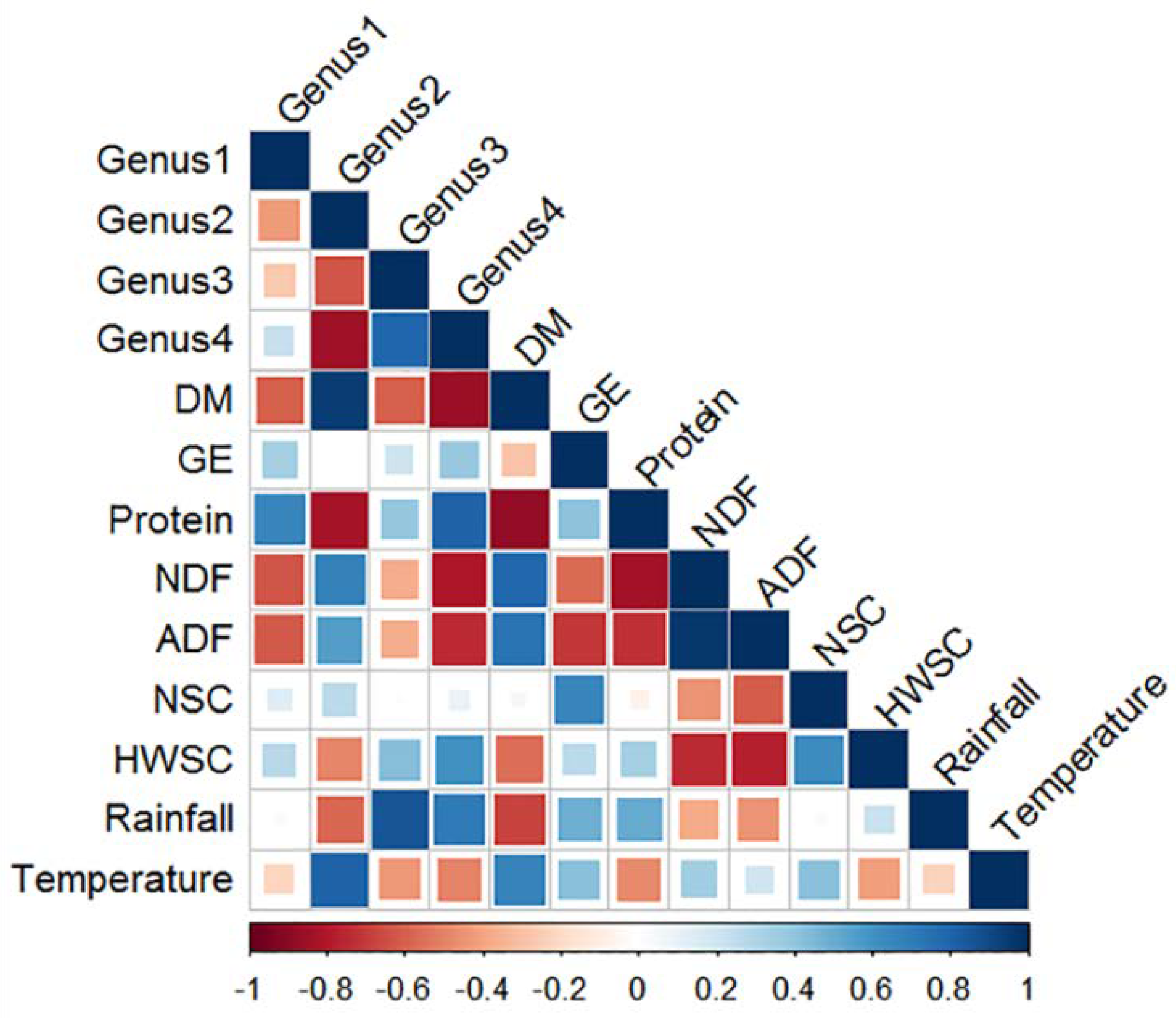
3.3.5. Correlation between Nutrient Composition, Climate Variables and Relative Abundance of Faecal Bacterial Genera
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janis, C. The evolutionary strategy of the Equidae and the origins of rumen and cecal digestion. Evolution 1976, 30, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Clauss, M.; Schiele, K.; Ortmann, S.; Fritz, J.; Codron, D.; Hummel, J.; Kienzle, E. The effect of very low food intake on digestive physiology and forage digestibility in horses. J. Anim. Physiol. Anim. Nutr. 2014, 98, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadet-Bourgeteau, S.; Julliand, V. Equine Microbial Gastro-Intestinal Health; Ellis, A.D., Longland, A.C., Coenen, M., Miraglia, N., Eds.; EAAP Publications; Wageningen Academic Press: Wageningen, The Netherlands, 2010; Volume 128, pp. 161–182. [Google Scholar]
- Costa, M.C.; Weese, J.S. The equine intestinal microbiome. Anim. Health Res. Rev. 2012, 13, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.; Hastie, P.; Murray, J.A. Factors Influencing Equine Gut Microflora: Current Knowledge. J. Equine Vet. Sci. 2020, 88, 102943. [Google Scholar] [CrossRef]
- Daly, K.; Stewart, C.S.; Flint, H.J.; Shirazi-Beechey, S.P. Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiol. Ecol. 2001, 38, 141–151. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Kittelmann, S.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Bermingham, E.N.; Thomas, D.G. Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 2014, 9, e112846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougal, K.; de la Fuente, G.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Newbold, C.J. Identification of a core bacterial community within the large intestine of the horse. PLoS ONE 2013, 8, e77660. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, M.M.; Harris, H.M.B.; Jeffery, I.B.; Claesson, M.J.; Younge, B.; O’Toole, P.W.; Ross, R. The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett. Appl. Microbiol. 2013, 57, 492–501. [Google Scholar] [CrossRef]
- Costa, M.C.; Arroyo, L.G.; Allen-Vercoe, E.; Stampfli, H.R.; Kim, P.T.; Sturgeon, A.; Weese, J.S. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3–V5 region of the 16S rRNA gene. PLoS ONE 2012, 7, e41484. [Google Scholar] [CrossRef] [Green Version]
- Vörös, A. Diet Related Changes in the Gastrointestinal Microbiota of Horses. Master’s Thesis, Swedish University of Agricultural Sciences Uppsala, Uppsala, Sweden, 2008. [Google Scholar]
- Dougal, K.; de la Fuente, G.; Harris, P.A.; Girdwood, S.E.; Pinloche, E.; Geor, R.J.; Nielsen, B.D.; Schott, H.C.; Elzinga, S.; Newbold, C.J. Characterisation of the faecal bacterial community in adult and elderly horses fed a high fibre, high oil or high starch diet using 454 pyrosequencing. PLoS ONE 2014, 9, e87424. [Google Scholar] [CrossRef] [Green Version]
- Weese, J.S.; Holcombe, S.J.; Embertson, R.M.; Kurtz, K.A.; Roessner, H.A.; Jalali, M.; Wismer, S.E. Changes in the faecal microbiota of mares precede the development of post partum colic. Equine Vet. J. 2015, 47, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Moreau, M.M.; Eades, S.C.; Reinemeyer, C.R.; Fugaro, M.N.; Onishi, J.C. Illumina sequencing of the V4 hypervariable region 16S rRNA gene reveals extensive changes in bacterial communities in the cecum following carbohydrate oral infusion and development of early-stage acute laminitis in the horse. Vet. Microbiol. 2014, 168, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Milinovich, G.J.; Klieve, A.V.; Pollitt, C.C.; Trott, D.J. Microbial events in the hindgut during carbohydrate-induced equine laminitis. Vet. Clin. N. Am. Equine Pract. 2010, 26, 79–94. [Google Scholar] [CrossRef] [PubMed]
- USDA. Lameness and Laminitis in U.S. Horses; USDA:APHIS:VS; CEAH; National Animal Health Monitoring System: Fort Collins, CO, USA, 2000. Available online: https://www.aphis.usda.gov/animal_health/nahms/equine/downloads/equine98/Equine98_dr_Lameness.pdf (accessed on 1 February 2015).
- Hoskin, S.; Gee, E. Feeding value of pastures for horses. N. Z. Vet. J. 2004, 52, 332–341. [Google Scholar] [CrossRef]
- Rogers, C.W.; Gee, E.K.; Firth, E.C. A cross-sectional survey of Thoroughbred stud farm management in the North Island of New Zealand. N. Z. Vet. J. 2007, 55, 302–307. [Google Scholar] [CrossRef]
- Verhaar, N.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Rosanowski, S.M. The feeding practices and estimated workload in a cohort of New Zealand competition horses. J. Equine Vet. Sci. 2014, 34, 1257–1262. [Google Scholar] [CrossRef]
- Fernandes, K.A.; Rogers, C.W.; Gee, E.K.; Bolwell, C.F.; Thomas, D.G. A cross-sectional survey of rider and horse demographics, and the feeding, health and management of Pony Club horses in New Zealand. Proc. N. Z. Soc. Anim. Prod. 2014, 74, 11–16. [Google Scholar]
- Litherland, A.J.; Woodward, S.J.R.; Stevens, D.R.; McDougal, D.B.; Boom, C.J.; Knight, T.L.; Lambert, M.G. Seasonal variations in pasture quality on New Zealand sheep and beef farms. Proc. N. Z. Soc. Anim. Prod. 2002, 62, 138–142. [Google Scholar]
- Longland, A.C.; Byrd, B.M. Pasture nonstructural carbohydrates and equine laminitis. J. Nutr. 2006, 136, 2099S–2102S. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Koike, S.; Miyaji, M.; Hata, H.; Tanaka, K. Hindgut microbes, fermentation and their seasonal variations in Hokkaido native horses compared to light horses. Ecol. Res. 2006, 21, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Salem, S.E.; Maddox, T.W.; Berg, A.; Antczak, P.; Ketley, J.M.; Williams, N.J.; Archer, D.C. Variation in faecal microbiota in a group of horses managed on pasture over a 12-month period. Sci. Rep. 2018, 8, 8510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henneke, D.R.; Potter, G.D.; Kreider, J.L.; Yeates, B.F. Relationship between condition score, physical measurements and body fat percentage in mares. Equine Vet. J. 1983, 15, 371–372. [Google Scholar] [CrossRef]
- Carter, R.A.; Geor, R.J.; Staniar, W.B.; Cubitt, T.A.; Harris, P.A. Apparent adiposity assessed by standardised scoring systems and morphometric measurements in horses and ponies. Vet. J. 2009, 179, 204–210. [Google Scholar] [CrossRef]
- Pelletier, S.; Tremblay, G.F.; Bertrand, A.; Bélanger, G.; Castonguay, Y.; Michaud, R. Drying procedures affect non-structural carbohydrates and other nutritive value attributes in forage samples. Anim. Feed Sci. Technol. 2010, 157, 139–150. [Google Scholar] [CrossRef]
- Udén, P. The influence of sample preparation of forage crops and silages on recovery of soluble and non-structural carbohydrates and their predictions by Fourier transform mid-IR transmission spectroscopy. Anim. Feed Sci. Technol. 2010, 160, 49–61. [Google Scholar] [CrossRef]
- Rius, A.; Kittelmann, S.; Macdonald, K.; Waghorn, G.; Janssen, P.; Sikkema, E. Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture. J. Dairy Sci. 2012, 95, 5024–5034. [Google Scholar] [CrossRef] [Green Version]
- Kittelmann, S.; Pinares-Patiño, C.S.; Seedorf, H.; Kirk, M.R.; Ganesh, S.; McEwan, J.C.; Janssen, P.H. Two Different Bacterial Community Types Are Linked with the Low-Methane Emission Trait in Sheep. PLoS ONE 2014, 9, e103171. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Yang, B.; Wang, Y.; Qian, P.-Y. Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinform. 2016, 17, 135. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Morrison, M. Comparisons of Different Hypervariable Regions of rrs Genes for Use in Fingerprinting of Microbial Communities by PCR-Denaturing Gradient Gel Electrophoresis. Appl. Environ. Microbiol. 2004, 70, 4800–4806. [Google Scholar] [CrossRef] [Green Version]
- Bartram, A.K.; Lynch, M.D.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852. [Google Scholar] [CrossRef] [Green Version]
- Fadrosh, D.W.; Ma, B.; Gajer, P.; Sengamalay, N.; Ott, S.; Brotman, R.M.; Ravel, J. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2014, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Dumas, J. Procedes de I’analyse organique. Ann. Chim. Phys. 1831, 47, 198–205. [Google Scholar]
- Nelson, N. A photometric adaptation of the Somogyi method for the determination of glucose. J. Biol. Chem. 1944, 153, 375–380. [Google Scholar] [CrossRef]
- Somogyi, M. Notes on sugar determination. J. Biol. Chem. 1952, 195, 19–23. [Google Scholar] [CrossRef]
- Pagan, J. Measuring the digestible energy content of horse feeds. In Advances in Equine Nutrition; Pagan, J., Ed.; Nottingham University Press: Nottingham, UK, 1998; pp. 71–76. [Google Scholar]
- Hall, M. Challenges with nonfiber carbohydrate methods. J. Anim. Sci. 2003, 81, 3226–3232. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Cox, M.; Peterson, D.; Biggs, P. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2011, 6, 610–618. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Gihring, T.M.; Green, S.J.; Schadt, C.W. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 2012, 14, 285–290. [Google Scholar] [CrossRef]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.; Ryan, P. PAST: Palaeontological statistics software package for education and data analysis. Plalaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Spellerberg, I.F.; Fedor, P.J. A tribute to Claude Shannon (1916–2001) and a plea for more rigorous use of species richness, species diversity and the ‘Shannon–Wiener’ Index. Glob. Ecol. Biogeogr. 2003, 12, 177–179. [Google Scholar] [CrossRef] [Green Version]
- Gotelli, N.J.; Colwell, R.K. Estimating species richness. Biol. Divers. Front. Meas. Assess. 2011, 12, 39–54. [Google Scholar]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. Available online: http://www.nature.com/nmeth/journal/v9/n8/abs/nmeth.2066.html#supplementary-information (accessed on 1 February 2015). [CrossRef]
- Vazquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristoffersen, C.T. Diet Effects on the Short-Term Temporal Dynamics of the Equine Hindgut Microbiota. Master’s Thesis, Norwegian University of Life Sciences, Ås, Norway, 2014. [Google Scholar]
- Costa, M.; Stampfli, H.; Arroyo, L.; Allen-Vercoe, E.; Gomes, R.; Weese, J. Changes in the equine fecal microbiota associated with the use of systemic antimicrobial drugs. BMC Vet. Res. 2015, 11, 19. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.; Swetnam, L.D.; Turner, G.M.; Hancock, J.N.; Shearer, S.A. Storage Method Effects on Dry Matter and Quality Losses of Tall Fescue Round Bales. J. Prod. Agric. 1995, 8, 507–514. [Google Scholar] [CrossRef]
- Turner, J.E.; Coblentz, W.K.; Scarbrough, D.A.; Coffey, K.P.; Kellogg, D.W.; McBeth, L.J.; Rhein, R.T. Changes in Nutritive Value of Bermudagrass Hay during Storage. Agron. J. 2002, 94, 109–117. [Google Scholar] [CrossRef]
- Patel, D. Digestibility of FiberEzy and Timothy Haylage and Behavioural Observations and Voluntary Feed Intake of FiberEzy and Rye Clover Hay in Thoroughbred Horses. Master’s Thesis, Massey University, Palmerston North, New Zealand, 2014. [Google Scholar]
- Hirst, R.L. Seasonal Variation of Pasture Quality on Commercial Equine Farms in New Zealand. Master’s Thesis, Massey University, Palmerston North, New Zealand, 2011. [Google Scholar]
- Longland, A.; Cairns, A.; Humphreys, M. Seasonal and diurnal changes in fructan concentration in Lolium perenne: Implications for the grazing management of equine pre-disposed to laminitis. In Proceedings of the 16th Equine Nutrition and Physiology Symposium, Raleigh, NC, USA, 2–5 June 1999. [Google Scholar]
- Pearson, R.A.; Archibald, R.F.; Muirhead, R.H. A comparison of the effect of forage type and level of feeding on the digestibility and gastrointestinal mean retention time of dry forages given to cattle, sheep, ponies and donkeys. Br. J. Nutr. 2006, 95, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Pearson, R.A.; Merritt, J.B. Intake, digestion and gastrointestinal transit-time in resting donkeys and ponies and exercised donkeys given ad-libitum hay and straw diets. Equine Vet. J. 1991, 23, 339–343. [Google Scholar] [CrossRef]
- Faubladier, C.; Chaucheyras-Durand, F.; da Veiga, L.; Julliand, V. Effect of transportation on fecal bacterial communities and fermentative activities in horses: Impact of Saccharomyces cerevisiae CNCM I-1077 supplementation. J. Anim. Sci. 2013, 91, 1736–1744. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. Available online: http://www.nature.com/nature/journal/v486/n7402/abs/nature11053.html#supplementary-information (accessed on 1 February 2015). [CrossRef]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nutrient Composition | Diet | Apr | May | Jun | Jul | Aug | Sep | Oct | Nov | Dec | Jan | Feb | Mar |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DM (%) | P | 21.7 | 17.6 | 17.4 | 17.0 | 18.5 | 19.1 | 23.5 | 20.2 | 20.9 | 26.6 | 44.6 | 52.0 |
| H | 94.4 | 95.7 | 96.3 | 96.0 | 96.3 | ||||||||
| CP (%) | P | 21.0 | 24.2 | 26.9 | 27.5 | 24.7 | 21.9 | 25.5 | 21.1 | 28.2 | 19.9 | 15.1 | 13.2 |
| H | 15.0 | 9.3 | 11.1 | 8.8 | 9.8 | ||||||||
| Fat (%) | P | 3.2 | 3.6 | 3.8 | 3.4 | 3.3 | 3.6 | 3.9 | 3.7 | 4.4 | 3.3 | 2.3 | 2.1 |
| H | 1.4 | 1.4 | 1.4 | 1.5 | 1.5 | ||||||||
| CHO ‡ (%) | P | 67.0 | 58.5 | 58.1 | 55.0 | 54.7 | 61.1 | 65.7 | 70.5 | 61.3 | 72.4 | 77.6 | 79.7 |
| H | 76.2 | 82.4 | 79.2 | 81.9 | 80.4 | ||||||||
| Ash (%) | P | 13.0 | 17.8 | 15.3 | 17.8 | 20.5 | 18.0 | 10.1 | 9.7 | 11.2 | 10.0 | 10.2 | 9.2 |
| H | 11.0 | 8.6 | 9.1 | 8.9 | 9.2 | ||||||||
| HWSC (%) | P | 8.9 | 10.6 | 11.4 | 10.1 | 10.1 | 11.6 | 13.4 | 14.7 | 9.1 | 8.4 | 7.5 | 8.2 |
| H | 9.7 | 8.8 | 6.6 | 7.4 | 6.4 | ||||||||
| NDF (%) | P | 52.6 | 46.2 | 46.2 | 46.1 | 43.7 | 45.6 | 43.5 | 38.7 | 39.1 | 52.4 | 58.0 | 58.7 |
| H | 43.1 | 58.8 | 58.9 | 62.4 | 63.4 | ||||||||
| ADF (%) | P | 28.6 | 27.6 | 25.8 | 26.8 | 28.7 | 27.3 | 22.3 | 20.6 | 22.6 | 27.5 | 31.3 | 31.8 |
| H | 30.2 | 34.6 | 36.2 | 36.5 | 36.8 | ||||||||
| ADL (%) | P | 2.9 | 6.4 | 3.9 | 6.6 | 9.5 | 5.9 | 0.9 | 1.0 | 0.8 | 0.9 | 1.1 | 0.7 |
| H | 0.3 | 0.5 | 0.5 | 0.4 | 0.5 | ||||||||
| NSC § (%) | P | 10.1 | 8.2 | 7.8 | 5.2 | 7.8 | 10.8 | 17.0 | 26.8 | 17.1 | 14.5 | 14.4 | 16.8 |
| H | 29.5 | 21.9 | 19.5 | 18.4 | 16.3 | ||||||||
| GE (KJ/g) | P | 18.5 | 17.7 | 18.3 | 17.7 | 16.8 | 17.3 | 19.1 | 19.0 | 19.3 | 18.7 | 18.2 | 18.3 |
| H | 17.8 | 17.8 | 18.1 | 18.0 | 17.9 | ||||||||
| DE † (MJ/kg DM) | P | 8.1 | 7.8 | 8.3 | 10.0 | 10.8 | 10.2 | 9.2 | 8.5 | 8.7 | 8.5 | 8.2 | 8.6 |
| H | 9.8 | 8.8 | 8.6 | 8.3 | 8.1 | ||||||||
| Pasture variables | |||||||||||||
| Sward height (cm) | 3 | 4 | 4 | 4 | 4 | 4 | 5 | 7 | 7 | 6 | 4 | 4 | |
| Pasture cover (kg DM/ha) | 1290 | 1540 | 1300 | 1300 | 1300 | 1580 | 1780 | 2330 | 2500 | 2240 | 1710 | 1540 | |
| Climate variables | |||||||||||||
| Mean daily Rainfall (mm) | 3.7 | 1.1 | 2.8 | 1.8 | 1.9 | 4.6 | 3.9 | 2.1 | 2.2 | 1.9 | 0.8 | 0.4 | |
| Mean Temperature (max °C) | 20 | 16 | 14 | 14 | 15 | 16 | 18 | 21 | 22 | 22 | 24 | 22 |
| Details | Bacterial Sequences |
|---|---|
| Initial QC processed reads § | |
| Total number of reads | 16,075,202 |
| Mean number of reads per sample | 117,448 |
| (range) | (47,154–189,708) |
| High quality reads used in downstream analysis † | |
| Total number of reads | 5,257,753 |
| Mean number of reads per sample | 44,557 |
| (range) | (19,250–73,405) |
| Mean length of reads (bp) | 356 |
| (range) | (250–486) |
| Total OTUs detected at 97% similarity | 123,645 |
| Diversity Indices | Pasture | Pasture + Hay | p-Value | ||
|---|---|---|---|---|---|
| Median | IQR | Median | IQR | ||
| Simpson’s (diversity) | 0.90 | (0.89–0.91) | 0.89 | (0.88–0.90) | <0.001 * |
| Shannon-Weiner (entropy) | 3.02 | (2.93–3.12) | 2.91 | (2.84–2.96) | <0.001 * |
| Chao1 (richness) | 167 | (157–184) | 161 | (145–179) | 0.075 |
| Horse | Pasture | Pasture + Hay | ||
|---|---|---|---|---|
| Median | IQR | Median | IQR | |
| 1 | 0.21 | (0.17–0.25) | 0.20 | (0.17–0.24) |
| 2 | 0.18 | (0.14–0.20) | 0.18 | (0.15–0.21) |
| 3 | 0.19 | (0.16–0.24) | 0.23 | (0.19–0.26) |
| 4 | 0.21 | (0.16–0.25) | 0.23 | (0.18–0.29) |
| 5 | 0.21 | (0.16–0.28) | 0.18 | (0.16–0.24) |
| 6 | 0.23 | (0.19–0.28) | 0.20 | (0.18–0.24) |
| 7 | 0.19 | (0.15–0.22) | 0.17 | (0.14–0.21) |
| 8 | 0.22 | (0.20–0.24) | 0.21 | (0.16–0.23) |
| 9 | 0.21 | (0.18–0.27) | 0.22 | (0.21–0.26) |
| 10 | 0.22 | (0.18–0.28) | 0.24 | (0.19–0.27) |
| p value | <0.007 * | <0.001 * | ||
| Diversity Indices | Autumn | Winter | Summer | Spring | p-Value |
|---|---|---|---|---|---|
| Simpson’s (diversity) | 0.91 b | 0.89 a | 0.89 a | 0.89 a | 0.0001 |
| Shannon-Wiener (entropy) | 3.08 a | 2.93 bc | 3.00 a | 2.91 c | 0.0001 |
| Chao1 (richness) | 173 a | 169 a | 173 a | 154 b | 0.0002 |
| Diversity Indices | Apr | May | Jun | Jul | Aug | Sep | Oct | Nov | Dec | Jan | Feb | Mar | p-Value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Simpson’s (diversity) | 0.90 | 0.90 | 0.90 | 0.89 | 0.88 | 0.89 | 0.89 | 0.90 | 0.89 | 0.89 | 0.92 | 0.92 | 0.0001 |
| Shannon-Wiener (entropy) | 3.03 | 2.99 | 2.95 | 2.92 | 2.90 | 2.89 | 2.90 | 2.94 | 2.93 | 2.97 | 3.19 | 3.14 | 0.0001 |
| Chao1 (richness) | 162 | 169 | 171 | 187 | 142 | 148 | 165 | 144 | 162 | 162 | 192 | 181 | 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, K.A.; Gee, E.K.; Rogers, C.W.; Kittelmann, S.; Biggs, P.J.; Bermingham, E.N.; Bolwell, C.F.; Thomas, D.G. Seasonal Variation in the Faecal Microbiota of Mature Adult Horses Maintained on Pasture in New Zealand. Animals 2021, 11, 2300. https://doi.org/10.3390/ani11082300
Fernandes KA, Gee EK, Rogers CW, Kittelmann S, Biggs PJ, Bermingham EN, Bolwell CF, Thomas DG. Seasonal Variation in the Faecal Microbiota of Mature Adult Horses Maintained on Pasture in New Zealand. Animals. 2021; 11(8):2300. https://doi.org/10.3390/ani11082300
Chicago/Turabian StyleFernandes, Karlette A., Erica K. Gee, Chris W. Rogers, Sandra Kittelmann, Patrick J. Biggs, Emma N. Bermingham, Charlotte F. Bolwell, and David G. Thomas. 2021. "Seasonal Variation in the Faecal Microbiota of Mature Adult Horses Maintained on Pasture in New Zealand" Animals 11, no. 8: 2300. https://doi.org/10.3390/ani11082300
APA StyleFernandes, K. A., Gee, E. K., Rogers, C. W., Kittelmann, S., Biggs, P. J., Bermingham, E. N., Bolwell, C. F., & Thomas, D. G. (2021). Seasonal Variation in the Faecal Microbiota of Mature Adult Horses Maintained on Pasture in New Zealand. Animals, 11(8), 2300. https://doi.org/10.3390/ani11082300