
In Search of Species-Specific SNPs in a Non-Model Animal (European Bison (Bison bonasus))—Comparison of De Novo and Reference-Based Integrated Pipeline of STACKS Using Genotyping-by-Sequencing (GBS) Data



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Genomic DNA Extraction
2.2. Genotyping By-Sequencing Library Preparation and High-Throughput Sequencing
2.3. Processing of GBS RAD-Tags
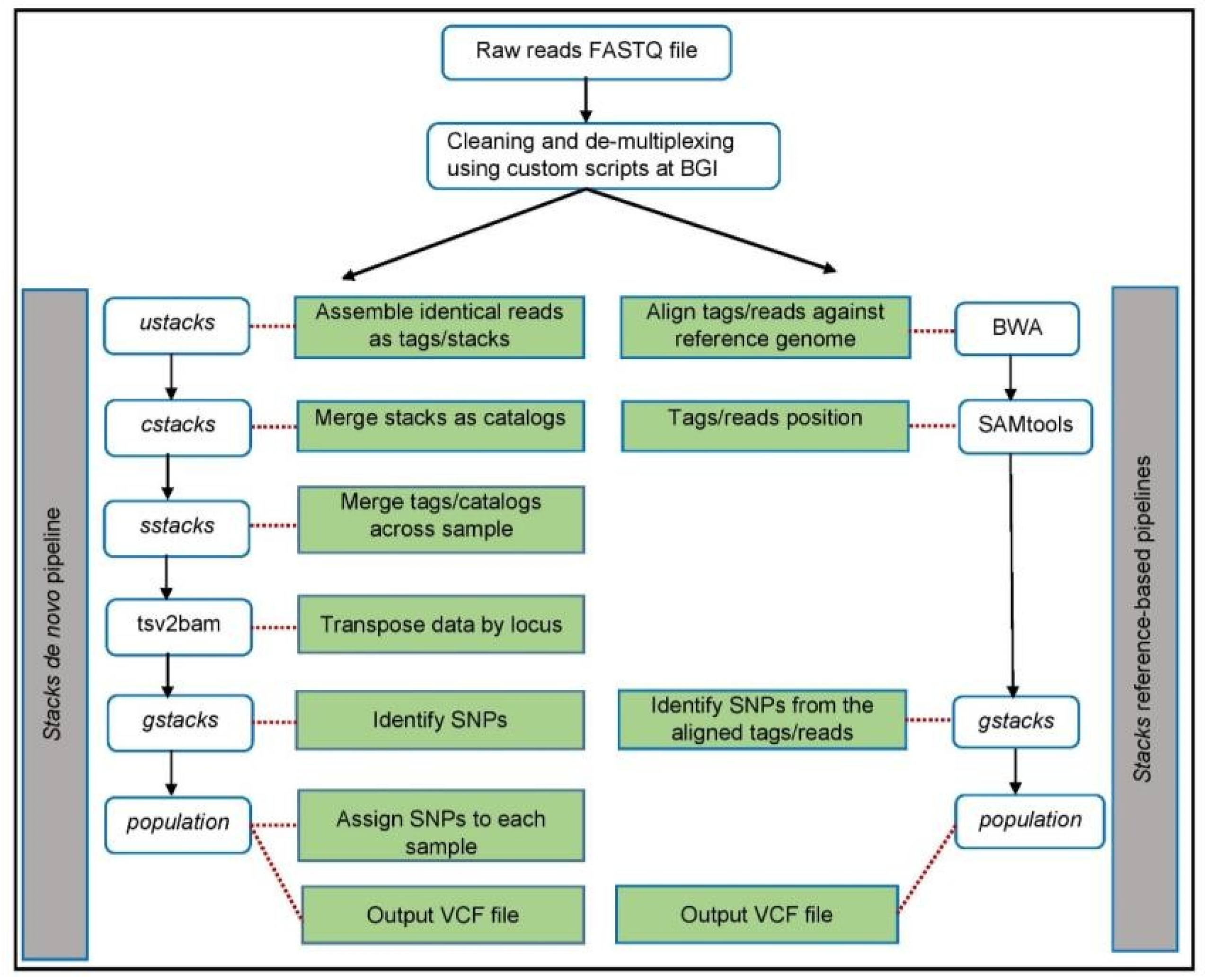
2.4. Bioinformatics Analyses
2.4.1. De Novo Pipeline
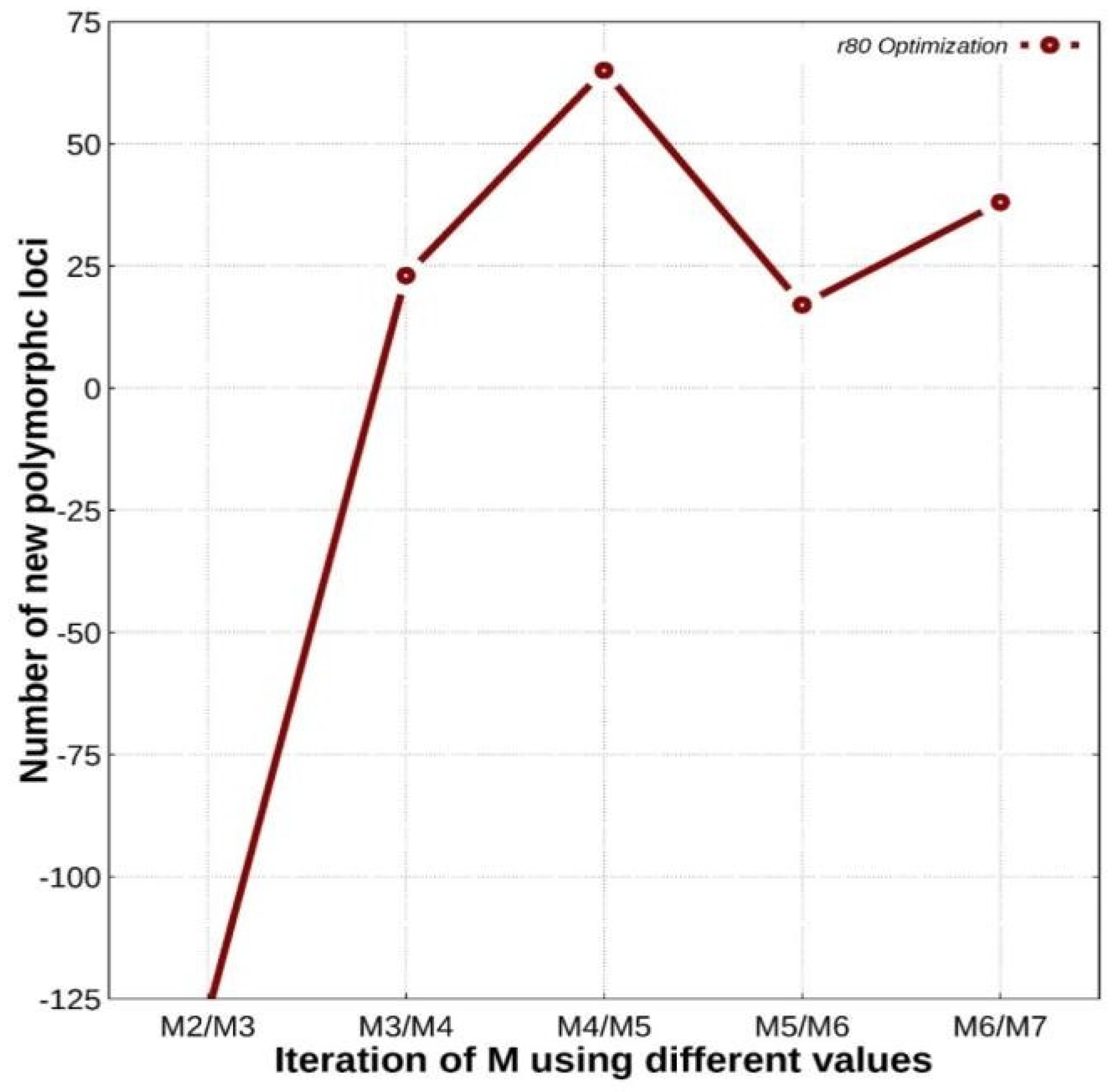
2.4.2. De Novo Parameter Optimization
2.4.3. Alignment and Variant Calling Using De Novo Pipeline (denovo_map.pl) of STACKS Software
2.4.4. Alignment and Variant Calling Using Reference Pipeline (ref_map.pl) of STACKS Software Using Bos taurus and the European Bison Genomes
2.4.5. Population Statistics
Measurements of Genetic Diversity
2.4.6. Variant Filtering
2.4.7. Juxtaposition of SNPs Obtained from De Novo Approach, Reference Pipeline, and Bovine High-Density SNP Chip Tool
3. Results
3.1. Sequence and Variant Calling
3.2. De Novo Parameter Optimization Results
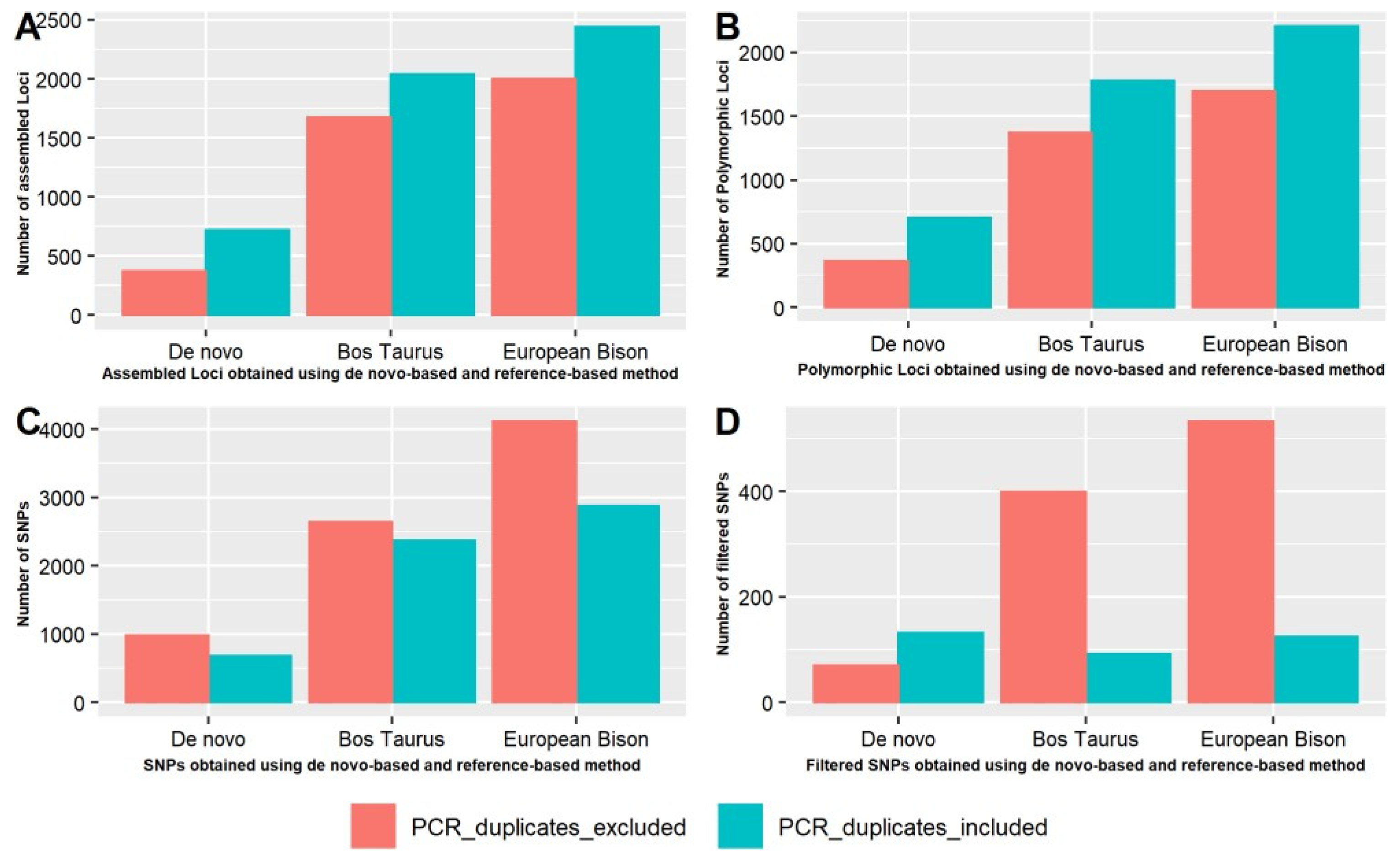
3.3. Comparison of Number of Catalogue Loci, Assembled Loci, Polymorphic Loci, and SNPs with and without PCR Duplicates Using De Novo Pipeline for European Bison Sequencing Data and Reference Pipeline for Bos taurus and European Bison
3.4. Population Statistics Results
3.5. Juxtaposition of SNPs Obtained from de novo Pipeline, Reference Pipelines, and Bovine High-Density SNP Chip Tool
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by- sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Alipour, H.; Bihamta, M.R.; Mohammadi, V.; Peyghambari, S.A.; Bai, G.; Zhang, G. Genotyping-by-Sequencing (GBS) Revealed Molecular Genetic Diversity of Iranian Wheat Landraces and Cultivars. Front. Plant Sci. 2017, 8, 1293. [Google Scholar] [CrossRef]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Legare, G.; Boyle, B.; Normandeau, É.; Laroche, J.; Larose, S.; Jean, M.; et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef] [Green Version]
- Hart, J.P.; Griffiths, P.D. Genotyping-by-Sequencing Enabled Mapping and Marker Development for the By-2 Potyvirus Resistance Allele in Common Bean. Plant Genome 2015, 8, eplantgenome2014090058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Donato, M.; Peters, S.O.; Mitchell, S.E.; Hussain, T.; Imumorin, I.G. Genotyping-by-sequencing (GBS): A novel, efficient and cost-effective genotyping method for cattle using next-generation sequencing. PLoS ONE 2013, 8, e62137. [Google Scholar] [CrossRef] [PubMed]
- Gurgul, A.; Miksza-Cybulska, A.; Szmatoła, T.; Semik-Gurgul, E.; Jasielczuk, I.; Bugno-Poniewierska, M.; Figarski, T.; Kajtoch, Ł. Evaluation of genotyping by sequencing for population genetics of sibling and hybridizing birds: An example using Syrian and Great Spotted Woodpeckers. J. Ornithol. 2018, 160, 287–294. [Google Scholar] [CrossRef]
- Zhu, F.; Cui, Q.Q.; Hou, Z.C. SNP discovery and genotyping using Genotyping-by-Sequencing in Pekin ducks. Sci. Rep. 2016, 6, 36223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.A.; Sharma, R.; Ahlawat, S.; Deb, R.; Negi, M.S.; Tripathi, S.B. Analysis of genetic relatedness among Indian cattle (Bos indicus) using genotyping-by-sequencing markers. Anim. Genet. 2018, 49, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Ashikari, M.; Jena, K.K.; Doi, K.; Reuscher, S. Adapting Genotyping-by-Sequencing for Rice F2 Populations. G3 (Bethesda) 2017, 7, 881–893. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.-B.; Yang, M.-H. Genotyping-by-Sequencing and Its Application to Oat Genomic Research. In Methods in Molecular Biology; Springer: Clifton, NJ, USA, 2017; Volume 1536, pp. 169–187. [Google Scholar] [CrossRef]
- Pértille, F.; Guerrero-Bosagna, C.; Silva VHd Boschiero, C.; Nunes, J.d.R.d.S.; Ledur, M.C.; Jensen, P.; Coutinho, L.L. High- Throughput and Cost-Effective Chicken Genotyping Using Next-Generation Sequencing. Sci. Rep. 2016, 6, 26929. Available online: http://europepmc.org/abstract/MED/27220827 (accessed on 19 October 2019). [CrossRef]
- Wang, Y.; Cao, X.; Zhao, Y.; Fei, J.; Hu, X.; Li, N. Optimized double-digest genotyping by sequencing (ddGBS) method with high-density SNP markers and high genotyping accuracy for chickens. PLoS ONE 2017, 12, e0179073. [Google Scholar] [CrossRef]
- Parker, C.C.; Gopalakrishnan, S.; Carbonetto, P.; Gonzales, N.M.; Leung, E.; Park, Y.J.; Aryee, E.; Davis, J.; Blizard, D.A.; Ackert- Bicknell, C.L.; et al. Genome-wide association study of behavioral, physiological and gene expression traits in outbred CFW mice. Nat. Genet. 2016, 48, 919–926. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Wittgenstein, H.; Mitchell, S.E.; Hyma, K.E.; Temnykh, S.V.; Kharlamova, A.V.; Gulevich, R.G.; Vladimirova, A.V.; Fong, H.W.F.; Acland, G.M.; et al. Genotyping-By-Sequencing (GBS) Detects Genetic Structure and Confirms Behavioral QTL in Tame and Aggressive Foxes (Vulpes vulpes). PLoS ONE 2015, 10, e0127013. [Google Scholar] [CrossRef] [Green Version]
- Etter, P.D.; Bassham, S.; Hohenlohe, P.A.; Johnson, E.A.; Cresko, W.A. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol. Biol. 2011, 772, 157–178. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Paris, J.R.; Stevens, J.R.; Catchen, J.M.; Johnston, S. Lost in parameter space: A road map for stacks. Methods Ecol. Evol. 2017, 8, 1360–1373. [Google Scholar] [CrossRef]
- Etter, P.D.; Preston, J.L.; Bassham, S.; Cresko, W.A.; Johnson, E.A. Local de novo assembly of RAD paired-end contigs using short sequencing reads. PLoS ONE 2011, 6, e18561. [Google Scholar] [CrossRef]
- Gore, M.A.; Chia, J.M.; Elshire, R.J.; Sun, Q.; Ersoz, E.S.; Hurwitz, B.L.; Peiffer, J.A.; McMullen, M.D.; Grills, G.S.; Ross-Ibarra, J.; et al. A first-generation haplotype map of maize. Science 2009, 326, 1115–1117. [Google Scholar] [CrossRef] [Green Version]
- Oleński, K.; Kamiński, S.; Tokarska, M.; Hering, D.M. Subset of SNPs for parental identification in European bison Lowland-Białowieża line (Bison bonasus bonasus). Conserv. Genet. Resour. 2017, 10, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Pertoldi, C.; Tokarska, M.; Wójcik, J.M.; Kawałko, A.; Randi, E.; Kristensen, T.N.; Loeschcke, V.; Coltman, D.; Wilson, G.A.; Gregersen, V.R.; et al. Phylogenetic relationships among the European and American bison and seven cattle breeds reconstructed using the BovineSNP50 Illumina Genotyping BeadChip. Acta Theriol. 2010, 55, 97–108. [Google Scholar] [CrossRef]
- Stronen, A.V.; Iacolina, L.; Pertoldi, C.; Tokarska, M.; Sørensen, B.S.; Bahrndorff, S.; Oleński, K.; Kamiński, S.; Nikolskiy, P. Genomic variability in the extinct steppe bison (Bison priscus) compared to the European bison (Bison bonasus). Mammal Res. 2018, 64, 127–131. [Google Scholar] [CrossRef]
- Olenski, K.; Tokarska, M.; Hering, D.; Puckowska, P.; Rusc, A.; Pertoldi, C.; Kamiński, S. Genome-wide association study for posthitis in the free-living population of European bison (Bison bonasus). Biol. Direct. 2015, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Pertoldi, C.; Wójcik, J.M.; Tokarska, M.; Kawałko, A.; Kristensen, T.N.; Loeschcke, V.; Gregersen, V.R.; Coltman, D.; Wilson, G.A.; Randi, E.; et al. Genome variability in European and American bison detected using the BovineSNP50 BeadChip. Conserv. Genet. 2009, 11, 627–634. [Google Scholar] [CrossRef]
- Tokarska, M.; Bunevich, A.N.; Demontis, D.; Sipko, T.; Perzanowski, K.; Baryshnikov, G.; Kowalczyk, R.; Voitukhovskaya, Y.; Wójcik, J.M.; Marczuk, B.; et al. Genes of the extinct Caucasian bison still roam the Białowieża Forest and are the source of genetic discrepances between Polish and Belarusian populations of the European bison, Bison bonasus. Biol. J. Linn. Soc. 2015, 114, 752–763. [Google Scholar] [CrossRef] [Green Version]
- Tokarska, M.; Marshall, T.; Kowalczyk, R.; Wojcik, J.M.; Pertoldi, C.; Kristensen, T.N.; Loeschcke, V.; Gregersen, V.R.; Bendixen, C. Effectiveness of microsatellite and SNP markers for parentage and identity analysis in species with low genetic diversity: The case of European bison. Heredity (Edinb) 2009, 103, 326–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertoldi, C.; Tokarska, M.; Wojcik, J.M.; Demontis, D.; Loeschcke, V.; Gregersen, V.R.; Coltman, D.; Wilson, G.A.; Randi, E.; Hansen, M.M.; et al. Depauperate genetic variability detected in the American and European bison using genomic techniques. Biol. Direct. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacolina, L.; Stronen, A.V.; Pertoldi, C.; Tokarska, M.; Nørgaard, L.S.; Muñoz, J.; Kjærsgaard, A.; Ruiz-Gonzalez, A.; Kamiński, S.; Purfield, D.C. Novel Graphical Analyses of Runs of Homozygosity among Species and Livestock Breeds. Biol. J. Linn. Soc. 2016, 114, 752–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McTavish, E.J.; Hillis, D.M. How do SNP ascertainment schemes and population demographics affect inferences about population history? BMC Genom. 2015, 16, 266. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Arce, N.; Rodriguez-Ezpeleta, N. Selecting RAD-Seq Data Analysis Parameters for Population Genetics: The More the Better? Front. Genet. 2019, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Fitz-Gibbon, S.; Hipp, A.L.; Pham, K.K.; Manos, P.S.; Sork, V.L. Phylogenomic inferences from reference-mapped and de novo assembled short-read sequence data using RADseq sequencing of California white oaks (Quercus section Quercus). Genome 2017, 60, 743–755. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- FASTQC. A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 19 October 2019).
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H. Stacks: Building and genotyping Loci de novo from short-read sequences. G3 (Bethesda) 2011, 1, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickland, D.P.; Battu, G.; Hudson, K.A.; Diers, B.W.; Hudson, M.E. A comparison of genotyping-by-sequencing analysis methods on low-coverage crop datasets shows advantages of a new workflow, GB-eaSy. BMC Bioinform. 2017, 18, 586. [Google Scholar] [CrossRef]
- Torkamaneh, D.; Laroche, J.; Belzile, F. Genome-Wide SNP Calling from Genotyping by Sequencing (GBS) Data: A Comparison of Seven Pipelines and Two Sequencing Technologies. PLoS ONE 2016, 11, e0161333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochette, N.C.; Catchen, J.M. Deriving genotypes from RAD-seq short-read data using Stacks. Nat. Protoc. 2017, 12, 2640–2659. [Google Scholar] [CrossRef]
- Mastretta-Yanes, A.; Arrigo, N.; Alvarez, N.; Jorgensen, T.H.; Pinero, D.; Emerson, B.C. Restriction site-associated DNA sequencing, genotyping error estimation and de novo assembly optimization for population genetic inference. Mol. Ecol. Resour. 2015, 15, 28–41. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Bassham, S.; Etter, P.D.; Stiffler, N.; Johnson, E.A.; Cresko, W.A. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010, 6, e1000862. [Google Scholar] [CrossRef] [Green Version]
- iGenomes Ready-To-Use Reference Sequences and Annotations. Available online: https://emea.support.illumina.com/sequencing/sequencing_software/igenome.html (accessed on 15 March 2019).
- Wang, K.; Wang, L.; Lenstra, J.A.; Jian, J.; Yang, Y.; Hu, Q.; Lai, D.; Qiu, Q.; Ma, T.; Du, Z.; et al. The genome sequence of the wisent (Bison bonasus). Gigascience 2017, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengjun, S.E.; Moran, V. Testing pipelines for genome-wide SNP calling from Genotyping-By-Sequencing (GBS) data for Pinus ponderosa. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Shafer, A.B.A.; Peart, C.R.; Tusso, S.; Maayan, I.; Brelsford, A.; Wheat, C.W.; Wolf, J.B. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol. Evol. 2016, 8, 907–917. [Google Scholar] [CrossRef]
- Ebbert, M.T.; Wadsworth, M.E.; Staley, L.A.; Hoyt, K.L.; Pickett, B.; Miller, J.; Duce, J.; Kauwe, J.S.; Ridge, P.G. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinform. 2016, 17 (Suppl. 7), 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarska, M.; Kawałko, A.; Wójcik, J.M.; Pertoldi, C. Genetic variability in the European bison (Bison bonasus) population from Białowieża forest over 50 years. Biol. J. Linn. Soc. 2009, 97, 801–809. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| B. taurus Reference: | E. Bison Reference: | De Novo | ||||
|---|---|---|---|---|---|---|
| PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | |
| Catalogue loci | 6,811,878 | 6,417,724 | 6,597,114 | 6,158,207 | 2,874,175 | 2,812,858 |
| Assembled loci | 2036 | 1676 | 2441 | 2001 | 721 | 374 |
| Poly. loci | 1779 | 1370 | 2205 | 1697 | 699 | 362 |
| SNPs 1 | 2371 | 2644 | 2878 | 4111 | 681 | 980 |
| SNPs 2 | 92 | 399 | 124 | 532 | 132 | 70 |
| De Novo/B. taurus | De Novo/E. Bison | |||
|---|---|---|---|---|
| PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | |
| Catalogue loci | 2,817,544 | 2,756,852 | 2,823,558 | 2,762,878 |
| Assembled loci | 717 | 371 | 719 | 372 |
| Poly. loci | 695 | 359 | 697 | 360 |
| SNPs 1 | 669 | 931 | 658 | 941 |
| SNPs 2 | 122 | 67 | 111 | 66 |
| B. taurus Reference: | E. Bison Reference: | De Novo | ||||
|---|---|---|---|---|---|---|
| PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | |
| All loci | ||||||
| %Poly.Loci | 0.57431 | 0.87891 | 0.54228 | 1.06225 | 0.50444 | 1.39282 |
| Obs_Het | 0.00028 | 0.00047 | 0.00029 | 0.00048 | 0.00074 | 0.00061 |
| Exp_Het | 0.00017 | 0.00065 | 0.00017 | 0.00068 | 0.00047 | 0.00055 |
| Fis | −0.00012 | 0.00174 | −0.00012 | 0.00195 | −0.00027 | −0.00012 |
| Var | 0.00031 | 0.00226 | 0.00034 | 0.00241 | 0.00089 | 0.00029 |
| StdErr | 0.01999 | 0.022 | 0.01857 | 0.02042 | 0.04156 | 0.03731 |
| Variant loci | ||||||
| Obs_Het | 0.04842 | 0.05308 | 0.0529 | 0.04529 | 0.14608 | 0.04369 |
| Exp_Het | 0.02969 | 0.0737 | 0.03146 | 0.06429 | 0.0922 | 0.03961 |
| Fis | −0.02122 | 0.19808 | −0.02303 | 0.18402 | −0.05428 | −0.00896 |
| Var | 0.05327 | 0.21865 | 0.06244 | 0.19384 | 0.17291 | 0.02092 |
| StdErr | 0.15438 | 0.20512 | 0.15109 | 0.15275 | 0.49407 | 0.28534 |
| B. taurus Reference | E. Bison Reference | ||||
|---|---|---|---|---|---|
| PCR dupl. | No PCR dupl. | PCR dupl. | No PCR dupl. | ||
| De novo SNPs | Common | 16 | 1 | 22 | 4 |
| Unique | 116 | 66 | 89 | 62 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunvar, S.; Czarnomska, S.; Pertoldi, C.; Tokarska, M. In Search of Species-Specific SNPs in a Non-Model Animal (European Bison (Bison bonasus))—Comparison of De Novo and Reference-Based Integrated Pipeline of STACKS Using Genotyping-by-Sequencing (GBS) Data. Animals 2021, 11, 2226. https://doi.org/10.3390/ani11082226
Kunvar S, Czarnomska S, Pertoldi C, Tokarska M. In Search of Species-Specific SNPs in a Non-Model Animal (European Bison (Bison bonasus))—Comparison of De Novo and Reference-Based Integrated Pipeline of STACKS Using Genotyping-by-Sequencing (GBS) Data. Animals. 2021; 11(8):2226. https://doi.org/10.3390/ani11082226
Chicago/Turabian StyleKunvar, Sazia, Sylwia Czarnomska, Cino Pertoldi, and Małgorzata Tokarska. 2021. "In Search of Species-Specific SNPs in a Non-Model Animal (European Bison (Bison bonasus))—Comparison of De Novo and Reference-Based Integrated Pipeline of STACKS Using Genotyping-by-Sequencing (GBS) Data" Animals 11, no. 8: 2226. https://doi.org/10.3390/ani11082226
APA StyleKunvar, S., Czarnomska, S., Pertoldi, C., & Tokarska, M. (2021). In Search of Species-Specific SNPs in a Non-Model Animal (European Bison (Bison bonasus))—Comparison of De Novo and Reference-Based Integrated Pipeline of STACKS Using Genotyping-by-Sequencing (GBS) Data. Animals, 11(8), 2226. https://doi.org/10.3390/ani11082226