
Detection of Genomic Regions with Pleiotropic Effects for Growth and Carcass Quality Traits in the Rubia Gallega Cattle Breed

 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Statistical Models
3. Results
3.1. Genetic Parameters
3.2. Genome-Wide Association Studies (GWAS)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gobierno de España, Ministerio de Agricultura Pesca y Alimentacion. Raza Bovina RUBIA GALLEGA. Available online: https://www.mapa.gob.es/es/ganaderia/temas/zootecnia/razas-ganaderas/razas/catalogo-razas/bovino/rubia-gallega/datos_morfologicos.aspx (accessed on 1 April 2021).
- Yagüe, G.; Goyache, F.; Becerra, J.; Moreno, C.; Sánchez, L.; Altarriba, J. Bayesian estimates of genetic parameters for pre-conception traits, gestation length and calving interval in beef cattle. Anim. Reprod. Sci. 2009, 114, 72–80. [Google Scholar] [CrossRef]
- Cantalapiedra, J.; Rodríguez, M.; Payán, R.; Camiña, M.; Iglesias, A. Evolución Morfológica de la Raza Rubia Gallega Basada en Sus Medidas Zoométricas e Índices Etnológicos. In Raças Autóctones no Espaço Ibérico: Um Recurso Sustentável; Marta-Costa, A.A., Tibério, M.L., Payan-Carreira, R., Eds.; Universidade de Trás-os-Montes e Alto Douro: Vila Real, Spain, 2016; pp. 65–69. Available online: https://www.ruralbit.com/client_manager/files/1448372206-1261.pdf (accessed on 15 March 2021).
- Legarra, A.; Christensen, O.F.; Aguilar, I.; Misztal, I. Single Step, a general approach for genomic selection. Livest. Sci. 2014, 166, 54–65. [Google Scholar] [CrossRef]
- Tiezzi, F.; Maltecca, C. Accounting for trait architecture in genomic predictions of US Holstein cattle using a weighted realized relationship matrix. Genet. Sel. Evol. 2015, 47, 24. [Google Scholar] [CrossRef]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef]
- Servin, B.; Stephens, M. Imputation-Based Analysis of Association Studies: Candidate Regions and Quantitative Traits. PLoS Genet. 2007, 3, e114. [Google Scholar] [CrossRef]
- Reglamento (CEE) n° 2237/91 del Consejo, de 26 de Julio de 1991, Por el Que se Modifica el Reglamento (CEE) n° 2930/81 Por el que se Establecen Disposiciones Complementarias Para la Aplicación del Modelo Comunitario de Clasificación de las Canales de Bovino. Available online: https://www.boe.es/doue/1991/204/L00011-00012.pdf (accessed on 25 March 2021).
- Ministerio de Agricultura, Pesca y Alimentación, B. Real Decreto 225/2008, de 15 de Febrero, Por el Que se Completa la Aplicación del Modelo Comunitario de Clasificación de las Canales de Vacuno Pesado y se Regula el Registro de los Precios de Mercado. Available online: https://www.boe.es/eli/es/rd/2008/02/15/225 (accessed on 25 March 2021).
- Chang, C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- VanRaden, P.M. Efficient Methods to Compute Genomic Predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, J.; Ding, X.; Bijma, P.; De Koning, D.-J.; Zhang, Q. Best Linear Unbiased Prediction of Genomic Breeding Values Using a Trait-Specific Marker-Derived Relationship Matrix. PLoS ONE 2010, 5, e12648. [Google Scholar] [CrossRef] [PubMed]
- Patterson, H.D.; Thompson, R. Recovery of Inter-Block Information when Block Sizes are Unequal. Biometrika 1971, 58, 545–554. [Google Scholar] [CrossRef]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs; University of Georgia: Athens, GA, USA, 2018. [Google Scholar]
- Aguilar, I.; Misztal, I.; Johnson, D.; Legarra, A.; Tsuruta, S.; Lawlor, T. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Savoia, S.; Albera, A.; Brugiapaglia, A.; Di Stasio, L.; Cecchinato, A.; Bittante, G. Heritability and genetic correlations of carcass and meat quality traits in Piemontese young bulls. Meat Sci. 2019, 156, 111–117. [Google Scholar] [CrossRef]
- Srivastava, S.; Lopez, B.I.; Heras-Saldana, S.D.L.; Park, J.-E.; Shin, D.-H.; Chai, H.-H.; Park, W.; Lee, S.-H.; Lim, D. Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle. Animal 2019, 9, 1061. [Google Scholar] [CrossRef]
- Altarriba, J.; Yagüe, G.; Moreno, C.; Varona, L. Exploring the possibilities of genetic improvement from traceability data. Livest. Sci. 2009, 125, 115–120. [Google Scholar] [CrossRef]
- Robinson, D. Estimation and interpretation of direct and maternal genetic parameters for weights of Australian Angus cattle. Livest. Prod. Sci. 1996, 45, 1–11. [Google Scholar] [CrossRef]
- Meyer, K. Estimates of genetic parameters for weaning weight of beef cattle accounting for direct-maternal environmental covariances. Livest. Prod. Sci. 1997, 52, 187–199. [Google Scholar] [CrossRef]
- Varona, L.; Munilla, S.; Casellas, J.; Moreno, C.; Altarriba, J. Consequences of paternally inherited effects on the genetic evaluation of maternal effects. Genet. Sel. Evol. 2015, 47, 63. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.; Näsholm, A.; Johansson, K.; Philipsson, J. Genetic analyses of field-recorded growth and carcass traits for Swedish beef cattle. Livest. Prod. Sci. 2003, 84, 53–62. [Google Scholar] [CrossRef]
- Purfield, D.; Evans, R.D.; Berry, D.P. Reaffirmation of known major genes and the identification of novel candidate genes associated with carcass-related metrics based on whole genome sequence within a large multi-breed cattle population. BMC Genom. 2019, 20, 720. [Google Scholar] [CrossRef]
- Grobet, L.; Martin, L.J.R.; Poncelet, D.; Pirottin, D.; Brouwers, B.; Riquet, J.; Schoeberlein, A.; Dunner, S.; Ménissier, F.; Massabanda, J.; et al. A deletion in the bovine myostatin gene causes the double–muscled phenotype in cattle. Nat. Genet. 1997, 17, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Bellinge, R.H.S.; Liberles, D.A.; Iaschi, S.P.A.; O’Brien, P.A.; Tay, G. Myostatinand its implications on animal breeding: A review. Anim. Genet. 2005, 36, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Taussat, S.; Vinet, A.; Krauss, D.; Maupetit, D.; Renand, G. Genetic parameters and genome-wide association study regarding feed efficiency and slaughter traits in Charolais cows. J. Anim. Sci. 2019, 97, 3684–3698. [Google Scholar] [CrossRef]
- Wiener, P.; Burton, D.; Ajmone-Marsan, P.; Dunner, S.; Mommens, G.; Nijman, I.; Rodellar, C.; Valentini, A.; Williams, J.L. Signatures of selection? Patterns of microsatellite diversity on a chromosome containing a selected locus. Heredity 2003, 90, 350–358. [Google Scholar] [CrossRef][Green Version]
- Rodriguez, A.G.; Munilla, S.; Mouresan, E.F.; Cañas-Álvarez, J.J.; Baro, J.A.; Molina, A.; Díaz, C.; Altarriba, J.; Piedrafita, J.; Varona, L. Genomic differentiation between Asturiana de los Valles, Avileña-Negra Ibérica, Bruna dels Pirineus, Morucha, Pirenaica, Retinta and Rubia Gallega cattle breeds. Animal 2017, 11, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Gil, B.; Arranz, J.J.; Wiener, P. An interpretive review of selective sweep studies in Bos taurus cattle populations: Identification of unique and shared selection signals across breeds. Front. Genet. 2015, 6, 6. [Google Scholar] [CrossRef]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef]
- Jin, J.; Arias, E.E.; Chen, J.; Harper, J.W.; Walter, J.C. A Family of Diverse Cul4-Ddb1-Interacting Proteins Includes Cdt2, which Is Required for S Phase Destruction of the Replication Factor Cdt1. Mol. Cell 2006, 23, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, Y.; Yu, B.; Wang, F.; Mi, T.; Zhao, Y. Silencing non-SMC chromosome-associated polypeptide G inhibits proliferation and induces apoptosis in hepatocellular carcinoma cells. Can. J. Physiol. Pharmacol. 2018, 96, 1246–1254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, J.; Guo, Y.; Zhang, L.; Xu, L.; Gao, X.; Zhu, B.; Gao, H.; Ni, H.; Chen, Y. Multi-strategy genome-wide association studies identify the DCAF16-NCAPG region as a susceptibility locus for average daily gain in cattle. Sci. Rep. 2016, 6, 38073. [Google Scholar] [CrossRef]
- Saatchi, M.; Schnabel, R.D.; Taylor, J.F.; Garrick, D.J. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genom. 2014, 15, 442. [Google Scholar] [CrossRef]
- Han, Y.J.; Chen, Y.; Liu, Y. Sequence variants of the LCORL gene and its association with growth and carcass traits in Qinchuan cattle in China. J. Genet. 2017, 96, 9–17. [Google Scholar] [CrossRef]
- Hoshiba, H.; Setoguchi, K.; Watanabe, T.; Kinoshita, A.; Mizoshita, K.; Sugimoto, Y.; Takasuga, A. Comparison of the effects explained by variations in the bovinePLAG1andNCAPGgenes on daily body weight gain, linear skeletal measurements and carcass traits in Japanese Black steers from a progeny testing program. Anim. Sci. J. 2013, 84, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Buaban, S.; Duangjinda, M.; Suzuki, M.; Masuda, Y.; Sanpote, J.; Kuchida, K. Short communication: Genetic analysis for fertility traits of heifers and cows from smallholder dairy farms in a tropical environment. J. Dairy Sci. 2015, 98, 4990–4998. [Google Scholar] [CrossRef]
- Pegolo, S.; Cecchinato, A.; Savoia, S.; Di Stasio, L.; Pauciullo, A.; Brugiapaglia, A.; Bittante, G.; Albera, A. Genome-wide association and pathway analysis of carcass and meat quality traits in Piemontese young bulls. Animal 2020, 14, 243–252. [Google Scholar] [CrossRef]
- Bhuiyan, M.S.A.; Lim, D.; Park, M.; Lee, S.; Kim, Y.; Gondro, C.; Park, B.; Lee, S. Functional Partitioning of Genomic Variance and Genome-Wide Association Study for Carcass Traits in Korean Hanwoo Cattle Using Imputed Sequence Level SNP Data. Front. Genet. 2018, 9, 217. [Google Scholar] [CrossRef]
- Lindholm-Perry, A.K.; Sexten, A.K.; A Kuehn, L.; Smith, T.P.; King, D.A.; Shackelford, S.D.; Wheeler, T.L.; Ferrell, C.L.; Jenkins, T.G.; Snelling, W.M.; et al. Association, effects and validation of polymorphisms within the NCAPG-LCORL locus located on BTA6 with feed intake, gain, meat and carcass traits in beef cattle. BMC Genet. 2011, 12, 103. [Google Scholar] [CrossRef]
- Kremer, B.E.; Adang, L.A.; Macara, I.G. Septins Regulate Actin Organization and Cell-Cycle Arrest through Nuclear Accumulation of NCK Mediated by SOCS7. Cell 2007, 130, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Seabury, C.M.; Oldeschulte, D.L.; Saatchi, M.; Beever, J.E.; Decker, J.E.; Halley, Y.A.; Bhattarai, E.K.; Molaei, M.; Freetly, H.C.; Hansen, S.L.; et al. Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle. BMC Genom. 2017, 18, 386. [Google Scholar] [CrossRef]
- Hu, T.; Krejsgaard, T.; Nastasi, C.; Buus, T.B.; Nansen, A.; Hald, A.; Spee, P.; Nielsen, P.R.; Blümel, E.; Gluud, M.; et al. Expression of the Voltage-Gated Potassium Channel Kv1.3 in Lesional Skin from Patients with Cutaneous T-Cell Lymphoma and Benign Dermatitis. Dermatology 2019, 236, 123–132. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Y.; Kim, Y.-S.; Iqbal, A.; Kim, J.-J. A whole genome association study to detect additive and dominant single nucleotide polymorphisms for growth and carcass traits in Korean native cattle, Hanwoo. Asian-Australas. J. Anim. Sci. 2016, 30, 8–19. [Google Scholar] [CrossRef]
- Jiménez-Martínez, M.; Stamatakis, K.; Fresno, M. The Dual-Specificity Phosphatase 10 (DUSP10): Its Role in Cancer, Inflammation, and Immunity. Int. J. Mol. Sci. 2019, 20, 1626. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, V.M.P.; Gouveia, G.C.; de Moraes, M.M.; de Araújo, A.E.M.; Raidan, F.S.S.; Fonseca, P.A.D.S.; Cardoso, E.P.; da Silva, M.V.G.B.; Toral, F.L.B. Genes underlying genetic correlation between growth, reproductive and parasite burden traits in beef cattle. Livest. Sci. 2021, 244, 104332. [Google Scholar] [CrossRef]
- Chang, T.; Xia, J.; Xu, L.; Wang, X.; Zhu, B.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. A genome-wide association study suggests several novel candidate genes for carcass traits in Chinese Simmental beef cattle. Anim. Genet. 2018, 49, 312–316. [Google Scholar] [CrossRef]
- Kajiho, H.; Saito, K.; Tsujita, K.; Kontani, K.; Araki, Y.; Kurosu, H.; Katada, T. RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 2003, 116, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Sakurai, K.; Minoda, T.; Yoshikawa, M.; Nakagawa, S.; Fukushima, S.; Kontani, K.; Katada, T. Characterization of RIN3 as a Guanine Nucleotide Exchange Factor for the Rab5 Subfamily GTPase Rab31. J. Biol. Chem. 2011, 286, 24364–24373. [Google Scholar] [CrossRef]
- Stronen, A.V.; Pertoldi, C.; Iacolina, L.; Kadarmideen, H.N.; Kristensen, T.N. Genomic analyses suggest adaptive differentiation of northern European native cattle breeds. Evol. Appl. 2019, 12, 1096–1113. [Google Scholar] [CrossRef]
- Dall, E.; Brandstetter, H. Mechanistic and structural studies on legumain explain its zymogenicity, distinct activation pathways, and regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 10940–10945. [Google Scholar] [CrossRef]
- Cai, H.; Li, M.; Sun, X.; Plath, M.; Li, C.; Lan, X.; Lei, C.; Huang, Y.; Bai, Y.; Qi, X.; et al. Global Transcriptome Analysis during Adipogenic Differentiation and Involvement of Transthyretin Gene in Adipogenesis in Cattle. Front. Genet. 2018, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Bolormaa, S.; Pryce, J.; Reverter-Gomez, T.; Zhang, Y.; Barendse, W.; Kemper, K.; Tier, B.; Savin, K.; Hayes, B.J.; Goddard, M.E. A Multi-Trait, Meta-analysis for Detecting Pleiotropic Polymorphisms for Stature, Fatness and Reproduction in Beef Cattle. PLoS Genet. 2014, 10, e1004198. [Google Scholar] [CrossRef]
- Zhang, Q.; Guldbrandtsen, B.; Thomasen, J.R.; Lund, M.S.; Sahana, G. Genome-wide association study for longevity with whole-genome sequencing in 3 cattle breeds. J. Dairy Sci. 2016, 99, 7289–7298. [Google Scholar] [CrossRef]
- Fang, Z.-H.; Pausch, H. Multi-trait meta-analyses reveal 25 quantitative trait loci for economically important traits in Brown Swiss cattle. BMC Genom. 2019, 20, 695. [Google Scholar] [CrossRef]
- Karaman, E.; Lund, M.S.; Su, G. Multi-trait single-step genomic prediction accounting for heterogeneous (co)variances over the genome. Heredity 2020, 124, 274–287. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Trait | Records | Mean | SD |
|---|---|---|---|
| Birth Weight, kg (BW) | 327,350 | 42.60 | 7.24 |
| Weaning Weight, kg (WW) | 83,818 | 284.63 | 47.49 |
| Cold Carcass Weight, kg (CCW) | 91,621 | 223.47 | 40.38 |
| Fatness, % (FAT) 1 | 91,475 | 2.27 | 0.44 |
| Conformation (CON) 2 | 91,609 | 3.95 | 0.66 |
| BW | WW | CCW | FAT | CON | |
|---|---|---|---|---|---|
| 12.70 (0.12) | 398.04 (7.75) | 182.27 (3.83) | 0.012 (0.5 × 10−3) | 0.033 (0.1 × 10−2) | |
| 10.89 (0.19) | 894.79 (54.48) | 724.43 (15.89) | 0.078 (0.2 × 10−2) | 0.219 (0.5 × 10−2) | |
| - | 301.97 (21.96) | - | - | - | |
| - | −382.72 (30.85) | - | - | - | |
| 23.68 (0.13) | 639.95 (26.36) | 224.16 (10.04) | 0.098 (0.2 × 10−2) | 0.136 (0.3 × 10−2) | |
| 0.230 | 0.400 | 0.641 | 0.415 | 0.565 | |
| - | 0.135 | - | - | - | |
| - | −0.736 | - | - | - |
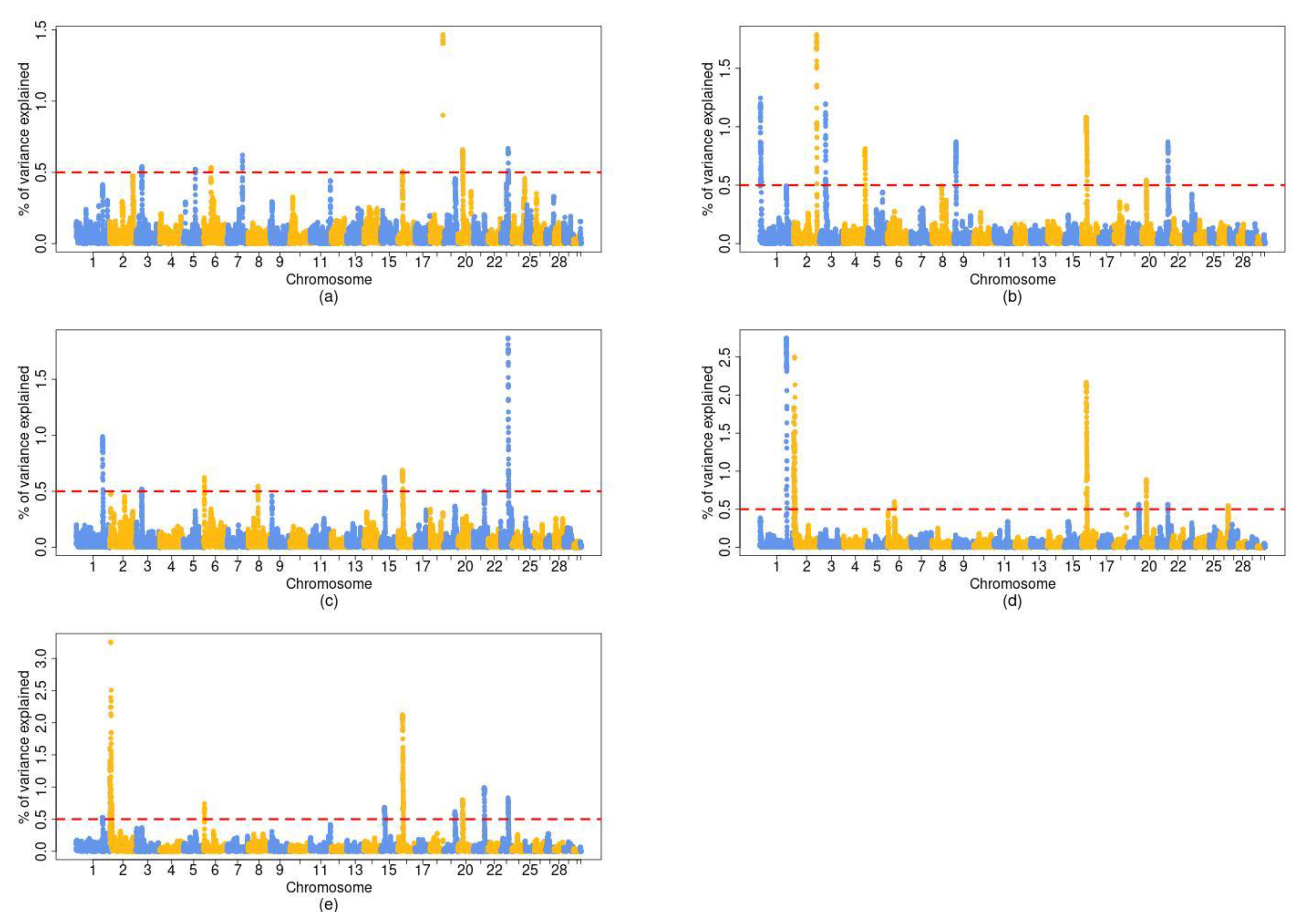
| Chromosome | BP 2 | BW | WW | CCW | FAT | CON | CG 3 |
|---|---|---|---|---|---|---|---|
| 1 | 131–132 | - | X | X | X | X | NCK1 |
| 2 | 1–11 | - | - | - | X | X | MSTN |
| 3 | 32–33 | X | X | X | - | - | KCNA3 |
| 6 | 36–38 | X | - | X | X | X | DCAF16, LCORL, NCAPG, LAP3, FAM184B |
| 15 | 23–24 | - | - | X | - | X | - |
| 16 | 25–26 | X | X | X | X | X | DUSP10 |
| 20 | 21–22 | X | X | - | X | X | - |
| 21 | 56–57 | - | X | - | X | X | RIN3, LGMN |
| 23 | 38–39 | X | - | X | - | X | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez-Castillero, M.; Then, C.; Altarriba, J.; Srihi, H.; López-Carbonell, D.; Díaz, C.; Martinez, P.; Hermida, M.; Varona, L. Detection of Genomic Regions with Pleiotropic Effects for Growth and Carcass Quality Traits in the Rubia Gallega Cattle Breed. Animals 2021, 11, 1682. https://doi.org/10.3390/ani11061682
Martinez-Castillero M, Then C, Altarriba J, Srihi H, López-Carbonell D, Díaz C, Martinez P, Hermida M, Varona L. Detection of Genomic Regions with Pleiotropic Effects for Growth and Carcass Quality Traits in the Rubia Gallega Cattle Breed. Animals. 2021; 11(6):1682. https://doi.org/10.3390/ani11061682
Chicago/Turabian StyleMartinez-Castillero, Maria, Carlos Then, Juan Altarriba, Houssemeddine Srihi, David López-Carbonell, Clara Díaz, Paulino Martinez, Miguel Hermida, and Luis Varona. 2021. "Detection of Genomic Regions with Pleiotropic Effects for Growth and Carcass Quality Traits in the Rubia Gallega Cattle Breed" Animals 11, no. 6: 1682. https://doi.org/10.3390/ani11061682
APA StyleMartinez-Castillero, M., Then, C., Altarriba, J., Srihi, H., López-Carbonell, D., Díaz, C., Martinez, P., Hermida, M., & Varona, L. (2021). Detection of Genomic Regions with Pleiotropic Effects for Growth and Carcass Quality Traits in the Rubia Gallega Cattle Breed. Animals, 11(6), 1682. https://doi.org/10.3390/ani11061682