
Whole-Genome Sequence Analysis of Italian Honeybees (Apis mellifera)

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
- -
- 61 Ligustica honeybees (A.m. ligustica) from Piedmont (2), Lombardy (6), Emilia-Romagna (23), Tuscany (11), Umbria (2), Marche (6), Abruzzo (8) and Puglia (3). We will refer to them as Ligustica.
- -
- 6 Sicilian bees (A. m. sicula). We will refer to them as Sicula.
- -
- 8 Carnica bees (A. m. carnica) from Lombardy (5), Piedmont, Trentino and Veneto (1 each). We will refer to them as Carnica.
- -
- 4 Mellifera bees (A.m. mellifera) along the western Riviera coast (Liguria). These samples actually represent a population located at the intersection of the Italian Ligustica and the French A.m. mellifera. Local beekeepers describe it as a typical stabilized hybrid well fitted to the local environment. We will refer to them as Mellifera.
- -
- 2 hybrid specimens Carnica by Ligustica (one in Lombardy and one in Trentino). We will refer to them as Carnica x Ligustica.
- -
- 43 Buckfast bees sampled in Lombardy (25), Veneto (2), Piedmont (15) and in Apulia (1). We will refer to them as Buckfast.
2.2. DNA Extraction
2.3. Library Preparation
2.4. Sequence Processing and Alignment
2.5. Single Nucleotide Polymorphism (SNP) Calling, Annotation and Filtering
2.6. SNP Analysis
3. Results
3.1. Sequencing and Read Mapping
3.2. Percentage of Specific and Common SNPs
3.3. Principal Component Analysis (PCA)
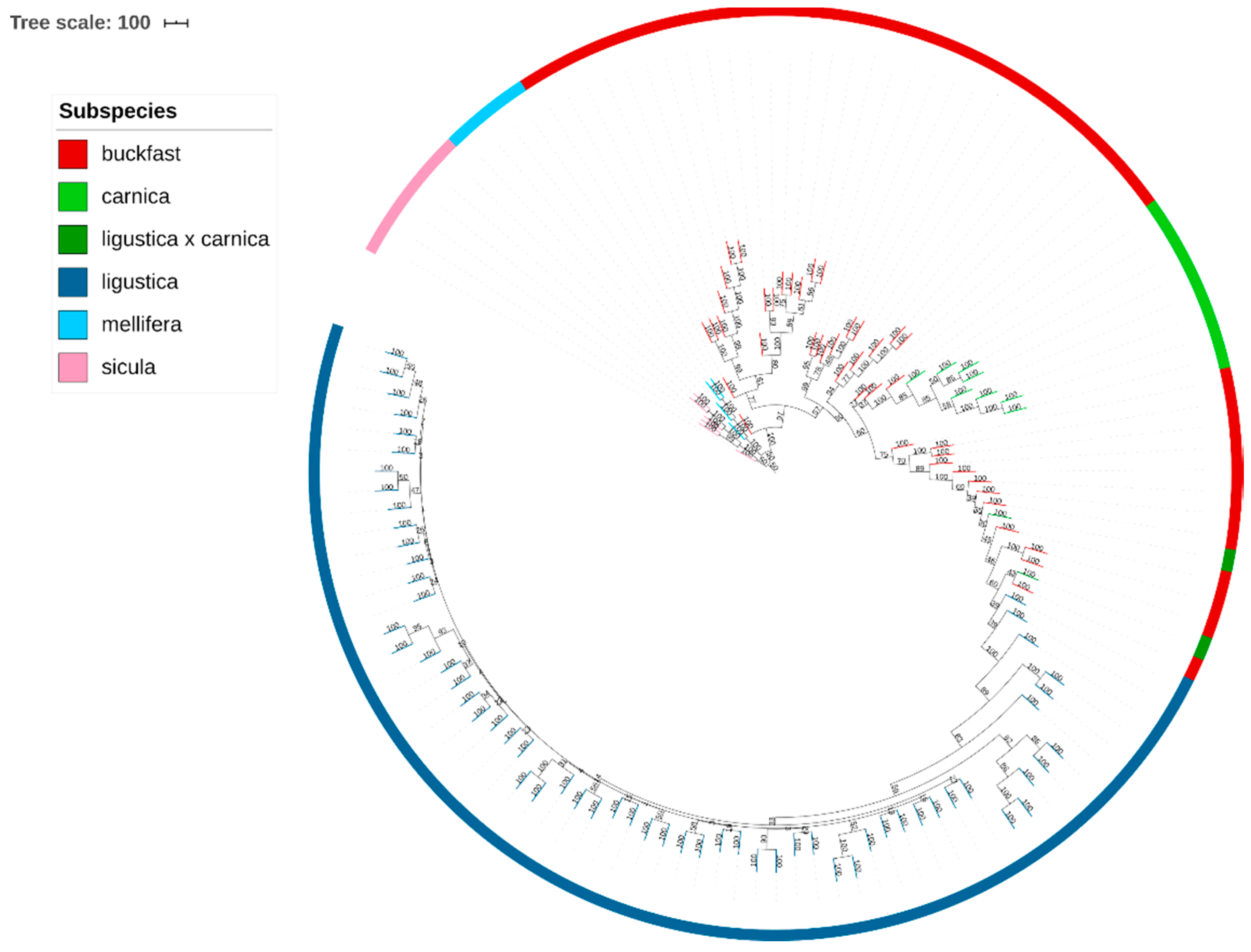
3.4. Phylogenetic Tree
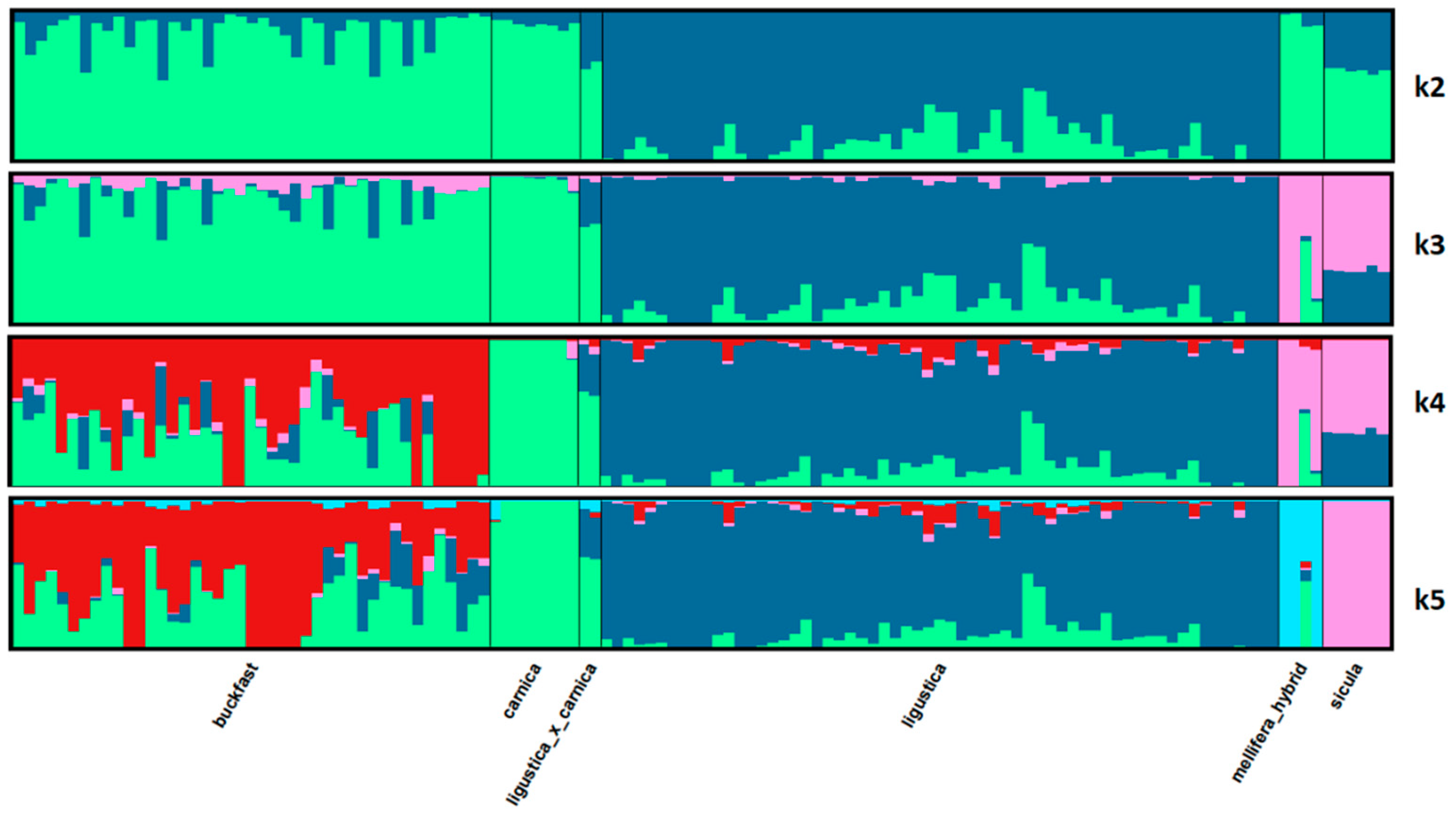
3.5. Admixture
3.6. Morphometric Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franck, P.; Garnery, L.; Celebrano, G.; Solignac, M.; Cornuet, J.M. Hybrid origins of honeybees from Italy (Apis mellifera ligustica) and Sicily (A. m. sicula). Mol. Ecol. 2000, 9, 907–921. [Google Scholar] [CrossRef]
- Ruttner, F. Biogeography and Taxonomy of Honeybees; Springer: Berlin/Heidelberg, Germany, 1988. [Google Scholar]
- Adam, B. Breeding the Honeybee; Northern Bee Books: Mytholmroyd, UK, 1987. [Google Scholar]
- Wallberg, A.; Han, F.; Wellhagen, G.; Dahle, B.; Kawata, M.; Haddad, N.; Simões, Z.L.P.; Allsopp, M.H.; Kandemir, I.; De La Rúa, P.; et al. Worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat. Genet. 2014, 46, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Parejo, M.; Wragg, D.; Gauthier, L.; Vignal, A.; Neumann, P.; Neuditschko, M. Using whole-genome sequence information to foster conservation effort for the European dark honey bee, apis mellifera mellifera. Front. Ecol. Evol. 2016. [Google Scholar] [CrossRef]
- Péntek-Zakar, E.; Oleksa, A.; Borowik, T.; Kusza, S. Population structure of honeybees in the Carpathian basin (Hungary) confirms introgression from surrounding subspecies. Ecol. Evol. 2015, 5, 5456–5467. [Google Scholar] [CrossRef]
- Wragg, D.; Techer, M.A.; Canale-Tabet, K.; Basso, B.; Bidanel, J.P.; Labarthe, E.; Bouchez, O.; Le Conte, Y.; Clémencet, J.; Delatte, H.; et al. Autosomal and mitochondrial adaptation following admixture: A case study on the honeybees of Reunion Island. Genome Biol. Evol. 2018, 10, 220–238. [Google Scholar] [CrossRef] [PubMed]
- Aminimoghadamfarouj, N.; Nematollahi, A. Structure elucidation and botanical characterization of diterpenes from a specific type of bee glue. Molecules 2017, 22, 1185. [Google Scholar] [CrossRef]
- Wragg, D.; Marimon, M.M.; Basso, B.; Bidanel, J.P.; Labarthe, E.; Bouchez, O.; Le Conte, Y.; Vignal, A. Whole-genome resequencing of honeybee drones to detect genomic selection in a population managed for royal jelly. Sci. Rep. 2016, 6, 27168. [Google Scholar] [CrossRef] [PubMed]
- Techer, M.A.; Clémencet, J.; Simiand, C.; Turpin, P.; Garnery, L.; Reynaud, B.; Delatte, H. Genetic diversity and differentiation among insular honey bee populations in the southwest Indian Ocean likely reflect old geographical isolation and modern introductions. PLoS ONE 2017, 12, e0189234. [Google Scholar] [CrossRef] [PubMed]
- Francoy, T.M.; Gonçalves, L.S.; De Jong, D. Rapid morphological changes in populations of hybrids between Africanized and European honey bees. Genet. Mol. Res. 2012, 11, 3349–3356. [Google Scholar] [CrossRef]
- Arias, M.C.; Sheppard, W.S. Molecular phylogenetics of honey bee subspecies (Apis mellifera L.) inferred from mitochondrial DNA sequence. Mol. Phylogenet. Evol. 1996, 5, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Dall’Olio, R.; Marino, A.; Lodesani, M.; Moritz, R.F.A. Genetic characterization of Italian honeybees, apis mellifera ligustica, based on microsatellite DNA polymorphism. Apidologie 2007, 38, 207–217. [Google Scholar] [CrossRef][Green Version]
- Rahimi, A.; Mirmoayedi, A.; Kahrizi, D.; Abdolshahi, R.; Kazemi, E.; Yari, K. Microsatellite genetic diversity of apis mellifera medaskorikov. Mol. Biol. Rep. 2014, 41, 7755–7761. [Google Scholar] [CrossRef]
- Il’yasov, R.A.; Poskryakov, A.V.; Petukhov, A.V.; Nikolenko, A.G. Genetic differentiation of local populations of the dark European bee apis mellifera mellifera. Genetika 2015, 51, 792–798. [Google Scholar] [CrossRef]
- Il’yasov, R.A.; Poskryakov, A.V.; Nikolenko, A.G. Seven genes of mitochondrial genome enabling differentiation of honeybee subspecies Apismellifera. Genetika 2016, 52, 1176–1184. [Google Scholar]
- Utzeri, V.J.; Ribani, A.; Fontanesi, L. Dispersion of Apis mellifera subspecies in the Emilia Romagna region (North of Italy) estimated using honey DNA analysis. Presented at 23rdASPA Congress Italy, Sorrento, Italy, 11–14 June 2019. [Google Scholar]
- Badino, G.; Celebrano, G.; Manino, A. Population structure and MDh-1 locus variation in apis mellifera ligustica. J. Hered. 1983, 74, 443–446. [Google Scholar] [CrossRef]
- Nazzi, F. Morphometric analysis of honeybees from an area racial hybridization in northeastern Italy. Apidologie 1992, 23, 89–96. [Google Scholar] [CrossRef]
- Parejo, M.; Wragg, D.; Henriques, D.; Charrière, J.D.; Estonba, A. Digging into the Genomic Past of Swiss Honey Bees by Whole-Genome Sequencing Museum Specimens. Genome Biol. Evol. 2020, 12, 2535–2551. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Zayed, A. Improving bee health through genomics. Nat. Rev. Genet. 2020, 21, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Bouga, M.; Alaux, C.; Bienkowska, M.; Büchler, R.; Carreck, N.L.; Cauia, E.; Chlebo, R.; Dahle, B.; Dall’Olio, R.; De la Rúa, P.; et al. A review of methods for discrimination of honey bee populations as applied to European beekeeping. J. Apic. Res. 2015, 50, 51–84. [Google Scholar] [CrossRef]
- Babraham Institute. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 2 September 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- SAMtools. Available online: http://samtools.sourceforge.net/ (accessed on 12 September 2020).
- Picard. Available online: https://broadinstitute.github.io/picard/ (accessed on 12 September 2020).
- Cornell University. Available online: https://arxiv.org/abs/1207.3907 (accessed on 12 September 2020).
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; De Pristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Kumar, H.; Panigrahi, M.; Chotaray, S.; Pal, D.; Bhanuprakash, V.; Saravanan, K.A.; Shandilya, R.; Parida, S.; Bhushan, B. Identification of breed-specific SNP panel in nine different cattle genomes. Biomed. Res. 2019, 30, 78–81. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bende, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Mass General Brigham. Available online: http://pngu.mgh.harvard.edu/purcell/plink/ (accessed on 12 September 2020).
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg Lab at Stanford University. Available online: http://web.stanford.edu/group/rosenberglab/distruct.html (accessed on 12 September 2020).
- Phylip. Available online: http://evolution.genetics.washington.edu/phylip.html (accessed on 12 September 2020).
- Interactive Tree of Life. Available online: https://itol.embl.de (accessed on 12 September 2020).
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Harpour, B.A.; Minaei, S.; Kent, C.F.; Zayed, A. Management increases genetic diversity of honey bees via admixture. Mol. Ecol. 2012, 21, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- FAO. Available online: http://www.fao.org/3/CA0103EN/ca0103en.pdf (accessed on 11 February 2020).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | % SNP | Number of Samples | N° SNP |
|---|---|---|---|
| A.m. ligustica | 64.19% | 61 | 2,811,524 |
| Buckfast | 69.76% | 43 | 3,055,491 |
| A.m. carnica | 46.67% | 8 | 2,044,148 |
| Hybridcarnica | 29.89% | 2 | 1,309,183 |
| A.m. mellifera | 49.74% | 4 | 2,178,614 |
| A.m. sicula | 59.68% | 6 | 2,613,986 |
| All | 124 | 4,380,004 |
| Ancestral Background (K) | CV Error |
|---|---|
| 2 | 0.206 |
| 3 | 0.210 |
| 4 | 0.214 |
| 5 | 0.223 |
| Clusters | ||||||
|---|---|---|---|---|---|---|
| Given Population | 1 | 2 | 3 | 4 | 5 | n |
| Buckfast | 0.018 | 0.318 | 0.083 | 0.010 | 0.572 | 43 |
| Carnica | 0.016 | 0.982 | 0.000 | 0.000 | 0.002 | 8 |
| Carnica × ligustica | 0.067 | 0.608 | 0.306 | 0.000 | 0.019 | 2 |
| Ligustica | 0.008 | 0.079 | 0.863 | 0.011 | 0.039 | 61 |
| Mellifera | 0.853 | 0.113 | 0.017 | 0.003 | 0.013 | 4 |
| Sicula | 0.000 | 0.000 | 0.000 | 1.000 | 0.000 | 6 |
| Population | A.m. mellifera | A.m. carnica | A.m. ligustica | A.m. sicula |
|---|---|---|---|---|
| A.m. mellifera | ||||
| A.m. carnica | 0.389 | |||
| A.m. ligustica | 0.383 | 0.153 | ||
| A.m. sicula | 0.308 | 0.259 | 0.207 | |
| Buckfast | 0.346 | 0.149 | 0.153 | 0.217 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minozzi, G.; Lazzari, B.; De Iorio, M.G.; Costa, C.; Carpana, E.; Crepaldi, P.; Rizzi, R.; Facchini, E.; Gandini, G.; Stella, A.; et al. Whole-Genome Sequence Analysis of Italian Honeybees (Apis mellifera). Animals 2021, 11, 1311. https://doi.org/10.3390/ani11051311
Minozzi G, Lazzari B, De Iorio MG, Costa C, Carpana E, Crepaldi P, Rizzi R, Facchini E, Gandini G, Stella A, et al. Whole-Genome Sequence Analysis of Italian Honeybees (Apis mellifera). Animals. 2021; 11(5):1311. https://doi.org/10.3390/ani11051311
Chicago/Turabian StyleMinozzi, Giulietta, Barbara Lazzari, Maria Grazia De Iorio, Cecilia Costa, Emanuele Carpana, Paola Crepaldi, Rita Rizzi, Elena Facchini, Gustavo Gandini, Alessandra Stella, and et al. 2021. "Whole-Genome Sequence Analysis of Italian Honeybees (Apis mellifera)" Animals 11, no. 5: 1311. https://doi.org/10.3390/ani11051311
APA StyleMinozzi, G., Lazzari, B., De Iorio, M. G., Costa, C., Carpana, E., Crepaldi, P., Rizzi, R., Facchini, E., Gandini, G., Stella, A., & Pagnacco, G. (2021). Whole-Genome Sequence Analysis of Italian Honeybees (Apis mellifera). Animals, 11(5), 1311. https://doi.org/10.3390/ani11051311