
Feline Coronavirus and Alpha-Herpesvirus Infections: Innate Immune Response and Immune Escape Mechanisms

 ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Immune Response
2.1. Pattern Recognition Receptors
2.2. Toll-Like Receptors
2.3. IFNs
2.4. Natural Killer Cells
2.5. MicroRNAs
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Pedersen, N.C. A review of feline infectious peritonitis virus infection: 1963–2008. J. Feline Med. Surg. 2009, 11, 225–258. [Google Scholar] [CrossRef] [PubMed]
- Bannasch, M.J.; Foley, J.E. Epidemiologic evaluation of multiple respiratory pathogens in cats in animal shelters. J. Feline Med. Surg. 2005, 7, 109–119. [Google Scholar] [CrossRef]
- Robert-Tissot, C.; Ruegger, V.L.; Cattori, V.; Meli, M.L.; Riond, B.; Moore, P.F.; Engels, M.; Franchini, M.; Hofmann-Lehmann, R.; Lutz, H. Stimulation with a class A CpG oligonucleotide enhances resistance to infection with feline viruses from five different families. Vet. Res. 2012, 43, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontier, D.; Fouchet, D.; Bahi-Jaber, N.; Poulet, H.; Guiserix, M.; Natoli, E.; Sauvage, F. When domestic cat (Felis silvestris catus) population structures interact with their viruses. C. R. Biol. 2009, 332, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Malbon, A.J.; Meli, M.L.; Barker, E.N.; Davidson, A.D.; Tasker, S.; Kipar, A. Inflammatory Mediators in the Mesenteric Lymph Nodes, Site of a Possible Intermediate Phase in the Immune Response to Feline Coronavirus and the Pathogenesis of Feline Infectious Peritonitis? J. Comp. Pathol. 2019, 166, 69–86. [Google Scholar] [CrossRef]
- Lee, S.; Channappanavar, R.; Kanneganti, T.D. Coronaviruses: Innate Immunity, Inflammasome Activation, Inflammatory Cell Death, and Cytokines. Trends Immunol. 2020, 41, 1083–1099. [Google Scholar] [CrossRef]
- Addie, D.D. Feline Coronavirus Infections. In Infectious Diseases of the Dog and Cat, 4th ed.; Greene, C.E., Ed.; Linda Duncan: St. Louis, MO, USA, 2012; pp. 92–108. [Google Scholar]
- ICTV-International Committee on Taxonomy of Viruses. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/dsdna-viruses/w/herpesviridae (accessed on 22 October 2021).
- Decaro, N.; Lorusso, A. Novel human coronavirus (SARS-CoV-2): A lesson from animal coronaviruses. Vet. Microbiol. 2020, 244, 108693. [Google Scholar] [CrossRef]
- Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; Lutz, H.; et al. Feline infectious peritonitis. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 594–604. [Google Scholar] [CrossRef]
- Pedersen, N.C. An update on feline infectious peritonitis: Diagnostics and therapeutics. Vet. J. 2014, 201, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Tekes, G.; Thiel, H.J. Feline Coronaviruses: Pathogenesis of Feline Infectious Peritonitis. Adv. Virus Res. 2016, 96, 193–218. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Boyle, J.F.; Floyd, K.; Fudge, A.; Barker, J. An enteric coronavirus infection of cats and its relationship to feline infectious peritonitis. Am. J. Vet. Res. 1981, 42, 368–377. [Google Scholar]
- Brown, M.A.; Troyer, J.L.; Pecon-Slattery, J.; Roelke, M.E.; O’Brien, S.J. Genetics and pathogenesis of feline infectious peritonitis virus. Emerg. Infect. Dis. 2009, 15, 1445–1452. [Google Scholar] [CrossRef]
- Kipar, A.; Meli, M.L. Feline infectious peritonitis: Still an enigma? Vet. Pathol. 2014, 51, 505–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, M.A. Feline Infectious Peritonitis: Update on Pathogenesis, Diagnostics, and Treatment. Vet. Clin. N. Am. Small Anim. Pract. 2020, 50, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Dye, C.; Siddell, S.G. Genomic RNA sequence of Feline coronavirus strain FIPV WSU-79/1146. J. Gen. Virol. 2005, 86, 2249–2253. [Google Scholar] [CrossRef]
- Haijema, B.J.; Volders, H.; Rottier, P.J. Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J. Virol. 2004, 78, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Dedeurwaerder, A.; Desmarets, L.M.; Olyslaegers, D.A.J.; Vermeulen, B.L.; Dewerchin, H.L.; Nauwynck, H.J. The role of accessory proteins in the replication of feline infectious peritonitis virus in peripheral blood monocytes. Vet. Microbiol. 2013, 162, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Gaskell, R.M.; Susan, D.; Alan, R. Feline Respiratory Disease. In Infectious Diseases of the Dog and Cat, 4th ed.; Greene, C.E., Ed.; Linda Duncan: St. Louis, MO, USA, 2012; pp. 151–162. [Google Scholar]
- Lewin, A.C.; Coghill, L.M.; McLellan, G.J.; Bentley, E.; Kousoulas, K.G. Genomic analysis for virulence determinants in feline herpesvirus type-1 isolates. Virus Genes 2020, 56, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Povey, R.C.; Johnson, R.H. A survey of feline viral rhinotracheitis and feline picornavirus infection in Britain. J. Small Anim. Pract. 1971, 12, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Maes, R. Felid herpesvirus type 1 infection in cats: A natural host model for alphaherpesvirus pathogenesis. ISRN Vet. Sci. 2012, 2012, 495830. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.R.; Maggs, D.J. Feline herpesvirus type-1 transcription is associated with increased nasal cytokine gene transcription in cats. Vet. Microbiol. 2005, 108, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Nelli, R.K.; Maes, R.; Kiupel, M.; Hussey, G.S. Use of a feline respiratory epithelial cell culture system grown at the air-liquid interface to characterize the innate immune response following feline herpesvirus 1 infection. Virus Res. 2016, 214, 39–48. [Google Scholar] [CrossRef]
- Thiry, E.; Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; et al. Feline herpesvirus infection. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 547–555. [Google Scholar] [CrossRef]
- Dawson, S.; Willoughby, K.; Gaskell, R.M.; Wood, G.; Chalmers, W.S. A field trial to assess the effect of vaccination against feline herpesvirus, feline calicivirus and feline panleucopenia virus in 6-week-old kittens. J. Feline Med. Surg. 2001, 3, 17–22. [Google Scholar] [CrossRef]
- Lee, Y.; Maes, R.; Tai, S.S.; Soboll Hussey, G. Viral replication and innate immunity of feline herpesvirus-1 virulence-associated genes in feline respiratory epithelial cells. Virus Res. 2019, 264, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.W. Innate Immunity. In Clinical Small Animal Internal Medicine; Bruyette, D., Ed.; Willey: Hoboken, NJ, USA, 2020; Volumn 1, pp. 421–422. [Google Scholar]
- Robert-Tissot, C.; Ruegger, V.L.; Cattori, V.; Meli, M.L.; Riond, B.; Gomes-Keller, M.A.; Vogtlin, A.; Wittig, B.; Juhls, C.; Hofmann-Lehmann, R.; et al. The innate antiviral immune system of the cat: Molecular tools for the measurement of its state of activation. Vet. Immunol. Immunopathol. 2011, 143, 269–281. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Majzoub, K.; Wrensch, F.; Baumert, T.F. The Innate Antiviral Response in Animals: An Evolutionary Perspective from Flagellates to Humans. Viruses 2019, 11, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.M.R. Innate Responses to Viral Infections. In Fields Virology, 7th ed.; Knipe, D.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volumn 1, pp. 189–213. [Google Scholar]
- Maclachlan, N.J.; Dubovi, E.J. Antiviral Immunity and Virus Vaccines. In Fenner’s Veterinary Virology, 5th ed.; MacLachlan, N.J., Dubovi, E.J., Eds.; Sara Tenney: London, UK, 2017; pp. 79–104. [Google Scholar]
- Wang, L.; Shen, H.; Zheng, Y.; Schumacher, L.; Li, G. Astrovirus in White-Tailed Deer, United States, 2018. Emerg. Infect. Dis. 2020, 26, 374–376. [Google Scholar] [CrossRef]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S.; Baker, D.L.; Baker, A. Cellular and Molecular Immunology, 9th ed.; Elsevier: Philadelphia, PA, USA, 2018; pp. 63–102. [Google Scholar]
- Turin, L.; Riva, F. Toll-like receptor family in domestic animal species. Crit. Rev. Immunol. 2008, 28, 513–538. [Google Scholar] [CrossRef]
- Chen, S.; Tian, J.; Li, Z.; Kang, H.; Zhang, J.; Huang, J.; Yin, H.; Hu, X.; Qu, L. Feline Infectious Peritonitis Virus Nsp5 Inhibits Type I Interferon Production by Cleaving NEMO at Multiple Sites. Viruses 2019, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Harty, J.T.; Varga, S.M. Toll-like receptor 4 deficiency increases disease and mortality after mouse hepatitis virus type 1 infection of susceptible C3H mice. J. Virol. 2009, 83, 8946–8956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Hickford, J.G.; Fang, Q.; Lin, Y.S. Allelic variation of the ovine Toll-like receptor 4 gene. Dev. Comp. Immunol. 2007, 31, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.; Veltrop, R.; Ikpeze, N.; Martin-Garcia, J.; Navas-Martin, S. Protective role of Toll-like Receptor 3-induced type I interferon in murine coronavirus infection of macrophages. Viruses 2012, 4, 901–923. [Google Scholar] [CrossRef]
- Birra, D.; Benucci, M.; Landolfi, L.; Merchionda, A.; Loi, G.; Amato, P.; Licata, G.; Quartuccio, L.; Triggiani, M.; Moscato, P. COVID 19: A clue from innate immunity. Immunol. Res. 2020, 68, 161–168. [Google Scholar] [CrossRef]
- Li, Y.; Chen, M.; Cao, H.; Zhu, Y.; Zheng, J.; Zhou, H. Extraordinary GU-rich single-strand RNA identified from SARS coronavirus contributes an excessive innate immune response. Microbes Infect. 2013, 15, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, J.; Kumar, A.; Zheng, M.; Atherton, S.S.; Yu, F.S. Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 2006, 117, 167–176. [Google Scholar] [CrossRef]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef] [Green Version]
- Gould, D. Feline herpesvirus-1: Ocular manifestations, diagnosis and treatment options. J. Feline Med. Surg. 2011, 13, 333–346. [Google Scholar] [CrossRef]
- Sergerie, Y.; Rivest, S.; Boivin, G. Tumor necrosis factor-alpha and interleukin-1 beta play a critical role in the resistance against lethal herpes simplex virus encephalitis. J. Infect. Dis. 2007, 196, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Wheat, W.; Chow, L.; Coy, J.; Contreras, E.; Lappin, M.; Dow, S. Activation of upper respiratory tract mucosal innate immune responses in cats by liposomal toll-like receptor ligand complexes delivered topically. J. Vet. Intern. Med. 2019, 33, 838–845. [Google Scholar] [CrossRef]
- Contreras, E.T.; Olea-Popelka, F.; Wheat, W.; Dow, S.; Hawley, J.; Lappin, M.R. Evaluation of liposome toll-like receptor ligand complexes for non-specific mucosal immunoprotection from feline herpesvirus-1 infection. J. Vet. Intern. Med. 2019, 33, 831–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J.; Valentine, R.C. Virus interference. II. Some properties of interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-lambda orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Dedeurwaerder, A.; Olyslaegers, D.A.J.; Desmarets, L.M.B.; Roukaerts, I.D.M.; Theuns, S.; Nauwynck, H.J. ORF7-encoded accessory protein 7a of feline infectious peritonitis virus as a counteragent against IFN-alpha-induced antiviral response. J. Gen. Virol. 2014, 95, 393–402. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Sen, G.C. Viral evasion of the interferon system. J. Interferon Cytokine Res. 2009, 29, 475–476. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, A.; Hashemi, V.; Shomali, N.; Asghari, F.; Gharibi, T.; Akbari, M.; Gholizadeh, S.; Jafari, A. Innate and adaptive immune responses against coronavirus. Biomed. Pharmacother. 2020, 132, 110859. [Google Scholar] [CrossRef]
- Chen, X.; Yang, X.; Zheng, Y.; Yang, Y.; Xing, Y.; Chen, Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein Cell 2014, 5, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Kindler, E.; Thiel, V. To sense or not to sense viral RNA--essentials of coronavirus innate immune evasion. Curr. Opin. Microbiol. 2014, 20, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Menachery, V.D.; Gralinski, L.E.; Mitchell, H.D.; Dinnon, K.H., 3rd; Leist, S.R.; Yount, B.L., Jr.; Graham, R.L.; McAnarney, E.T.; Stratton, K.G.; Cockrell, A.S.; et al. Middle East Respiratory Syndrome Coronavirus Nonstructural Protein 16 Is Necessary for Interferon Resistance and Viral Pathogenesis. mSphere 2017, 2, e00346-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, B.L.; Devriendt, B.; Olyslaegers, D.A.; Dedeurwaerder, A.; Desmarets, L.M.; Favoreel, H.W.; Dewerchin, H.L.; Nauwynck, H.J. Suppression of NK cells and regulatory T lymphocytes in cats naturally infected with feline infectious peritonitis virus. Vet. Microbiol. 2013, 164, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, H.L.; Cornelissen, E.; Van Hamme, E.; Smits, K.; Verhasselt, B.; Nauwynck, H.J. Surface-expressed viral proteins in feline infectious peritonitis virus-infected monocytes are internalized through a clathrin- and caveolae-independent pathway. J. Gen. Virol. 2008, 89, 2731–2740. [Google Scholar] [CrossRef]
- Cornelissen, E.; Dewerchin, H.L.; Van Hamme, E.; Nauwynck, H.J. Absence of surface expression of feline infectious peritonitis virus (FIPV) antigens on infected cells isolated from cats with FIP. Vet. Microbiol. 2007, 121, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, Y.; Liu, X.; Sun, X.; Zhang, J.; Qu, L. Feline Herpesvirus 1 US3 Blocks the Type I Interferon Signal Pathway by Targeting Interferon Regulatory Factor 3 Dimerization in a Kinase-Independent Manner. J. Virol. 2018, 92, e00047-18. [Google Scholar] [CrossRef] [Green Version]
- Li, S.F.; Zhao, F.R.; Shao, J.J.; Xie, Y.L.; Chang, H.Y.; Zhang, Y.G. Interferon-omega: Current status in clinical applications. Int. Immunopharmacol. 2017, 52, 253–260. [Google Scholar] [CrossRef]
- Ballin, A.C.; Schulz, B.; Helps, C.; Sauter-Louis, C.; Mueller, R.S.; Hartmann, K. Limited efficacy of topical recombinant feline interferon-omega for treatment of cats with acute upper respiratory viral disease. Vet. J. 2014, 202, 466–470. [Google Scholar] [CrossRef]
- Gil, S.; Leal, R.O.; Duarte, A.; McGahie, D.; Sepulveda, N.; Siborro, I.; Cravo, J.; Cartaxeiro, C.; Tavares, L.M. Relevance of feline interferon omega for clinical improvement and reduction of concurrent viral excretion in retrovirus infected cats from a rescue shelter. Res. Vet. Sci. 2013, 94, 753–763. [Google Scholar] [CrossRef]
- Ritz, S.; Egberink, H.; Hartmann, K. Effect of feline interferon-omega on the survival time and quality of life of cats with feline infectious peritonitis. J. Vet. Intern. Med. 2007, 21, 1193–1197. [Google Scholar] [CrossRef]
- Hartmann, K.; Binder, C.; Hirschberger, J.; Cole, D.; Reinacher, M.; Schroo, S.; Frost, J.; Egberink, H.; Lutz, H.; Hermanns, W. Comparison of different tests to diagnose feline infectious peritonitis. J. Vet. Intern. Med. 2003, 17, 781–790. [Google Scholar] [CrossRef]
- Haid, C.; Kaps, S.; Gonczi, E.; Hassig, M.; Metzler, A.; Spiess, B.M.; Richter, M. Pretreatment with feline interferon omega and the course of subsequent infection with feline herpesvirus in cats. Vet. Ophthalmol. 2007, 10, 278–284. [Google Scholar] [CrossRef]
- Zuo, W.; Zhao, X. Natural killer cells play an important role in virus infection control: Antiviral mechanism, subset expansion and clinical application. Clin. Immunol. 2021, 227, 108727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Z.; Huang, J.; Chen, S.; Yin, H.; Tian, J.; Qu, L. miR-101 inhibits feline herpesvirus 1 replication by targeting cellular suppressor of cytokine signaling 5 (SOCS5). Vet. Microbiol. 2020, 245, 108707. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Z.; Huang, J.; Yin, H.; Tian, J.; Qu, L. miR-26a Inhibits Feline Herpesvirus 1 Replication by Targeting SOCS5 and Promoting Type I Interferon Signaling. Viruses 2019, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Guo, X.K.; Gao, L.; Huang, C.; Li, N.; Jia, X.; Liu, W.; Feng, W.H. MicroRNA-23 inhibits PRRSV replication by directly targeting PRRSV RNA and possibly by upregulating type I interferons. Virology 2014, 450–451, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Heo, I.; Kim, V.N. Modifications of small RNAs and their associated proteins. Cell 2010, 143, 703–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldipur, B.; Bhukya, P.L.; Arankalle, V.; Lole, K. Positive Regulation of Hepatitis E Virus Replication by MicroRNA-122. J. Virol. 2018, 92, e01999-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, S.C.; Tate, M.D.; Hertzog, P.J. MicroRNA as Type I Interferon-Regulated Transcripts and Modulators of the Innate Immune Response. Front. Immunol. 2015, 6, 334. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Hou, J.; Lin, L.; Wang, C.; Liu, X.; Li, D.; Ma, F.; Wang, Z.; Cao, X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J. Immunol. 2010, 185, 6226–6233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wang, B.; Chen, X.; He, Z.; Wang, Y.; Li, X.; Cao, H.; Zheng, S.J. MicroRNA gga-miR-130b Suppresses Infectious Bursal Disease Virus Replication via Targeting of the Viral Genome and Cellular Suppressors of Cytokine Signaling 5. J. Virol. 2018, 92, e01646-17. [Google Scholar] [CrossRef] [Green Version]
 and
and  , respectively.
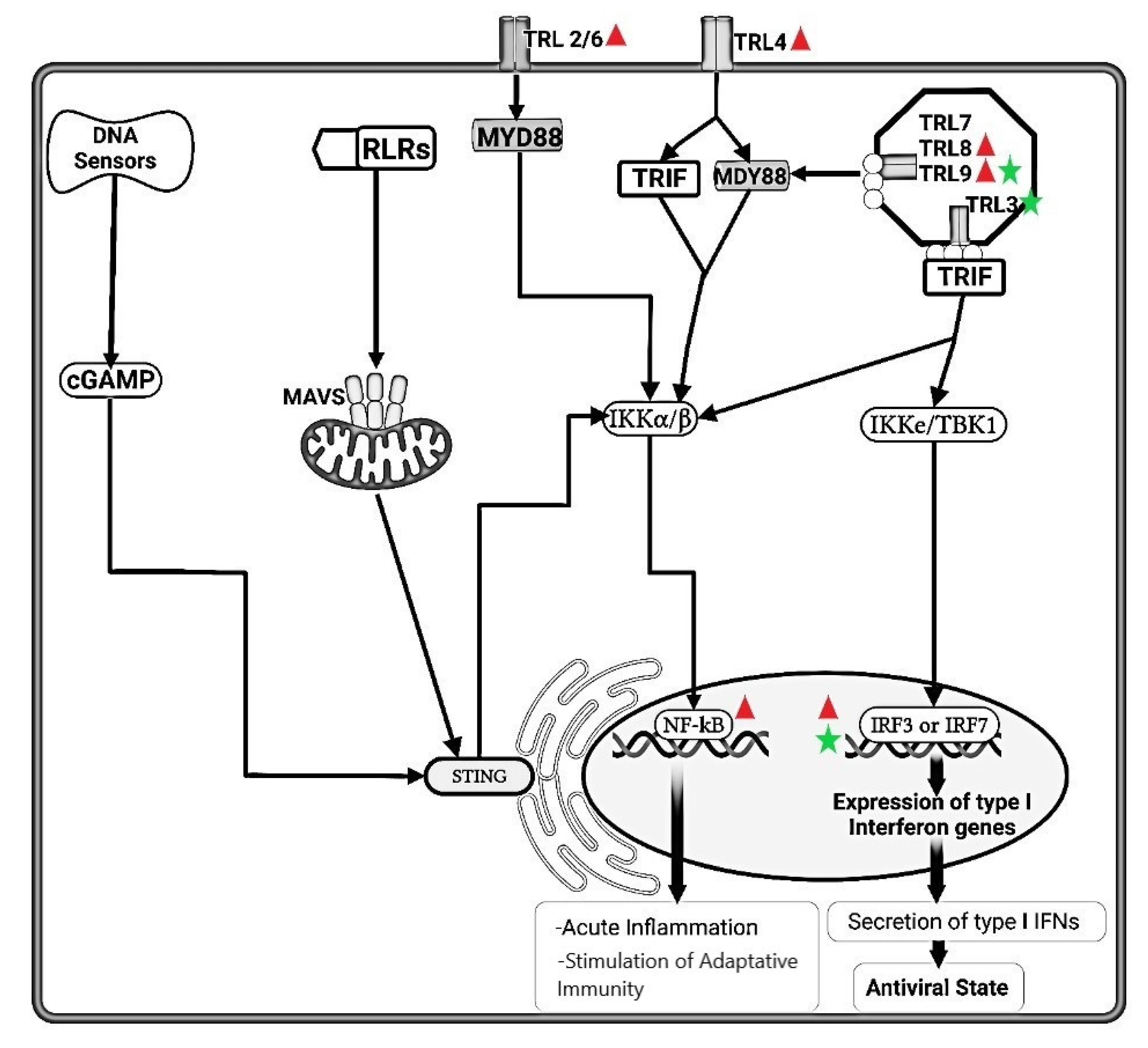
, respectively.  FeCoV infection induced higher gene expression level of TLRs 2, 4, 8 and 9, but not TLRs 3 and 7, suggesting either lack of an appropriate trigger, or virus inhibition of TLR trascripion. The synthesis of accessory proteins 7a and 3 by FeCoV are probably involved in the inhibition of type I IFN synthesis. The FIPV nsp5 produced an inhibition of IRF3 phosphorylation and suppression of type I IFN production.
FeCoV infection induced higher gene expression level of TLRs 2, 4, 8 and 9, but not TLRs 3 and 7, suggesting either lack of an appropriate trigger, or virus inhibition of TLR trascripion. The synthesis of accessory proteins 7a and 3 by FeCoV are probably involved in the inhibition of type I IFN synthesis. The FIPV nsp5 produced an inhibition of IRF3 phosphorylation and suppression of type I IFN production.  FeHV-1 infection induced an upregulation of TRL9 expression and a downregulation of TLR3. The FeHV-1 US3 protein competitively binds to IRF binding domain hindering dimerization of IRF3. TIR, Toll IL-1 receptor; TLR, Toll-like receptors; MyD88, Myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing interferon-β; NF-κB, Nuclear Factor kappa B; IRFs, Interferon Regulatory Factors; IFN, Interferon; IKK, Inhibitor-KbKinase; TBK1, TANK Binding Kinase 1; cGAMP, Cyclic guanosine monophosphate-adenosine monophosphate; STING, Stimulator of Interferon Genes; RLRs, RIG-I-like receptors; MAVs, Mitochondrial antiviral-signaling protein; nsp, nonstructural protein.
and , respectively. FeCoV infection induced higher gene expression level of TLRs 2, 4, 8 and 9, but not TLRs 3 and 7, suggesting either lack of an appropriate trigger, or virus inhibition of TLR trascripion. The synthesis of accessory proteins 7a and 3 by FeCoV are probably involved in the inhibition of type I IFN synthesis. The FIPV nsp5 produced an inhibition of IRF3 phosphorylation and suppression of type I IFN production. FeHV-1 infection induced an upregulation of TRL9 expression and a downregulation of TLR3. The FeHV-1 US3 protein competitively binds to IRF binding domain hindering dimerization of IRF3. TIR, Toll IL-1 receptor; TLR, Toll-like receptors; MyD88, Myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing interferon-β; NF-κB, Nuclear Factor kappa B; IRFs, Interferon Regulatory Factors; IFN, Interferon; IKK, Inhibitor-KbKinase; TBK1, TANK Binding Kinase 1; cGAMP, Cyclic guanosine monophosphate-adenosine monophosphate; STING, Stimulator of Interferon Genes; RLRs, RIG-I-like receptors; MAVs, Mitochondrial antiviral-signaling protein; nsp, nonstructural protein.
FeHV-1 infection induced an upregulation of TRL9 expression and a downregulation of TLR3. The FeHV-1 US3 protein competitively binds to IRF binding domain hindering dimerization of IRF3. TIR, Toll IL-1 receptor; TLR, Toll-like receptors; MyD88, Myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing interferon-β; NF-κB, Nuclear Factor kappa B; IRFs, Interferon Regulatory Factors; IFN, Interferon; IKK, Inhibitor-KbKinase; TBK1, TANK Binding Kinase 1; cGAMP, Cyclic guanosine monophosphate-adenosine monophosphate; STING, Stimulator of Interferon Genes; RLRs, RIG-I-like receptors; MAVs, Mitochondrial antiviral-signaling protein; nsp, nonstructural protein.
and , respectively. FeCoV infection induced higher gene expression level of TLRs 2, 4, 8 and 9, but not TLRs 3 and 7, suggesting either lack of an appropriate trigger, or virus inhibition of TLR trascripion. The synthesis of accessory proteins 7a and 3 by FeCoV are probably involved in the inhibition of type I IFN synthesis. The FIPV nsp5 produced an inhibition of IRF3 phosphorylation and suppression of type I IFN production. FeHV-1 infection induced an upregulation of TRL9 expression and a downregulation of TLR3. The FeHV-1 US3 protein competitively binds to IRF binding domain hindering dimerization of IRF3. TIR, Toll IL-1 receptor; TLR, Toll-like receptors; MyD88, Myeloid differentiation primary response 88; TRIF, TIR-domain-containing adapter-inducing interferon-β; NF-κB, Nuclear Factor kappa B; IRFs, Interferon Regulatory Factors; IFN, Interferon; IKK, Inhibitor-KbKinase; TBK1, TANK Binding Kinase 1; cGAMP, Cyclic guanosine monophosphate-adenosine monophosphate; STING, Stimulator of Interferon Genes; RLRs, RIG-I-like receptors; MAVs, Mitochondrial antiviral-signaling protein; nsp, nonstructural protein.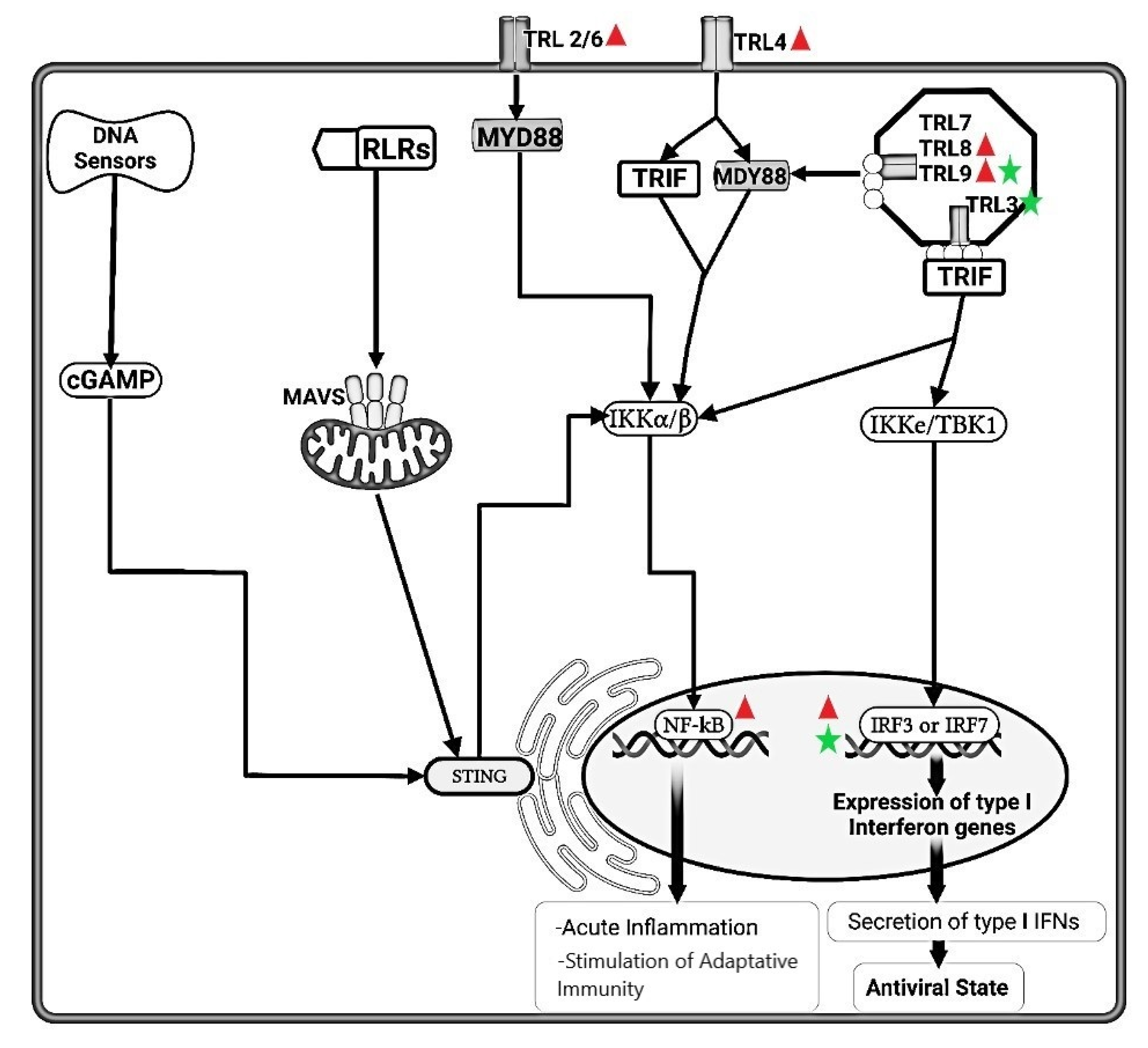
 .
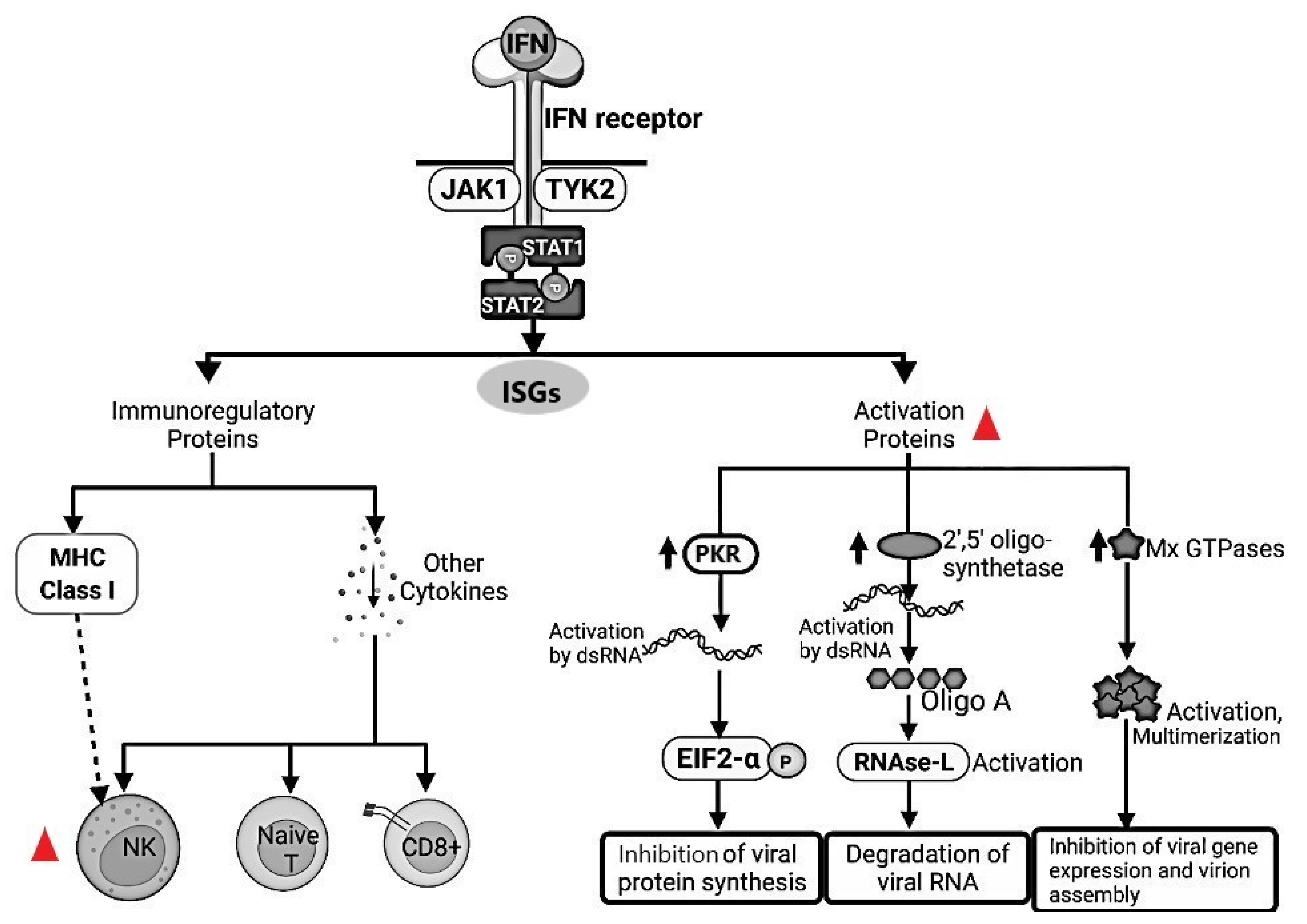
.  FeCoV infection inhibited the type I IFN synthesis that results in an inhibition of the protein synthesis. Natural Killer (NK) cells are drastically depleted from the peripheral blood mesenteric lymph nodes and spleen in FIPV-infected cats. Moreover, NK showed less cytotoxic activity in FIP-infected cats. dsRNA, Double-stranded RNA; PKR, Double-stranded RNA-activated protein kinase, EIF2-a, Eukaryotic translation initiation factor 2A; RNAse-L, Ribonuclease L; MHC, Major Histocompatibility Complex; NK, Natural Killer; Naive T, Naïve T cells; CD8+, Cytotoxic T lymphocytes; JAK1, Janus Kinase 1; TYK2, Tyrosine kinase 2; STAT1, Signal Transducer and Activator of Transcription 1; STAT2, Signal Transducer and Activator of Transcription 2; Mx GTPases, Mx dynamin-like GTPases; ISGs, Interferon stimulated genes.
. FeCoV infection inhibited the type I IFN synthesis that results in an inhibition of the protein synthesis. Natural Killer (NK) cells are drastically depleted from the peripheral blood mesenteric lymph nodes and spleen in FIPV-infected cats. Moreover, NK showed less cytotoxic activity in FIP-infected cats. dsRNA, Double-stranded RNA; PKR, Double-stranded RNA-activated protein kinase, EIF2-a, Eukaryotic translation initiation factor 2A; RNAse-L, Ribonuclease L; MHC, Major Histocompatibility Complex; NK, Natural Killer; Naive T, Naïve T cells; CD8+, Cytotoxic T lymphocytes; JAK1, Janus Kinase 1; TYK2, Tyrosine kinase 2; STAT1, Signal Transducer and Activator of Transcription 1; STAT2, Signal Transducer and Activator of Transcription 2; Mx GTPases, Mx dynamin-like GTPases; ISGs, Interferon stimulated genes.
FeCoV infection inhibited the type I IFN synthesis that results in an inhibition of the protein synthesis. Natural Killer (NK) cells are drastically depleted from the peripheral blood mesenteric lymph nodes and spleen in FIPV-infected cats. Moreover, NK showed less cytotoxic activity in FIP-infected cats. dsRNA, Double-stranded RNA; PKR, Double-stranded RNA-activated protein kinase, EIF2-a, Eukaryotic translation initiation factor 2A; RNAse-L, Ribonuclease L; MHC, Major Histocompatibility Complex; NK, Natural Killer; Naive T, Naïve T cells; CD8+, Cytotoxic T lymphocytes; JAK1, Janus Kinase 1; TYK2, Tyrosine kinase 2; STAT1, Signal Transducer and Activator of Transcription 1; STAT2, Signal Transducer and Activator of Transcription 2; Mx GTPases, Mx dynamin-like GTPases; ISGs, Interferon stimulated genes.
. FeCoV infection inhibited the type I IFN synthesis that results in an inhibition of the protein synthesis. Natural Killer (NK) cells are drastically depleted from the peripheral blood mesenteric lymph nodes and spleen in FIPV-infected cats. Moreover, NK showed less cytotoxic activity in FIP-infected cats. dsRNA, Double-stranded RNA; PKR, Double-stranded RNA-activated protein kinase, EIF2-a, Eukaryotic translation initiation factor 2A; RNAse-L, Ribonuclease L; MHC, Major Histocompatibility Complex; NK, Natural Killer; Naive T, Naïve T cells; CD8+, Cytotoxic T lymphocytes; JAK1, Janus Kinase 1; TYK2, Tyrosine kinase 2; STAT1, Signal Transducer and Activator of Transcription 1; STAT2, Signal Transducer and Activator of Transcription 2; Mx GTPases, Mx dynamin-like GTPases; ISGs, Interferon stimulated genes.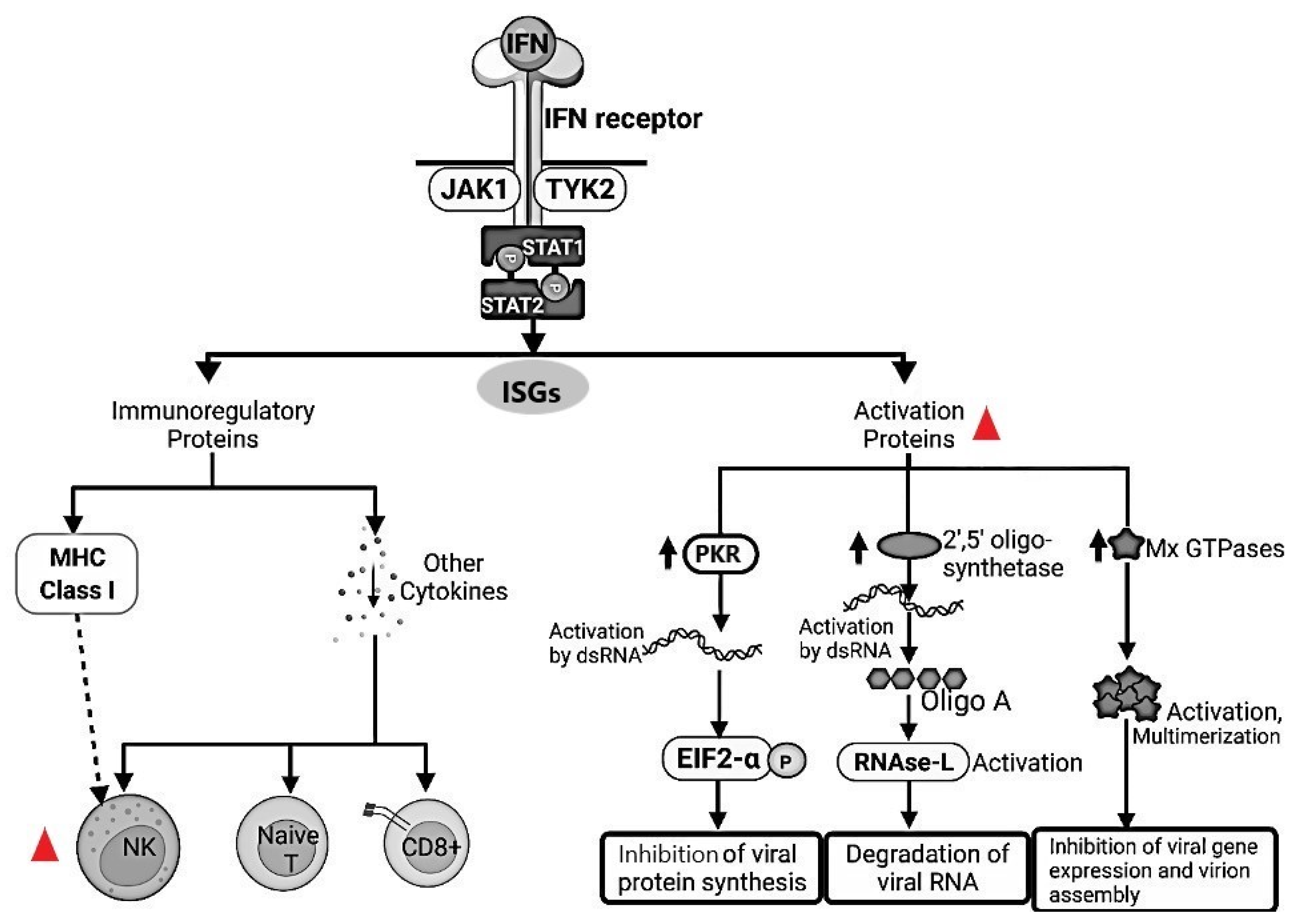
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capozza, P.; Pratelli, A.; Camero, M.; Lanave, G.; Greco, G.; Pellegrini, F.; Tempesta, M. Feline Coronavirus and Alpha-Herpesvirus Infections: Innate Immune Response and Immune Escape Mechanisms. Animals 2021, 11, 3548. https://doi.org/10.3390/ani11123548
Capozza P, Pratelli A, Camero M, Lanave G, Greco G, Pellegrini F, Tempesta M. Feline Coronavirus and Alpha-Herpesvirus Infections: Innate Immune Response and Immune Escape Mechanisms. Animals. 2021; 11(12):3548. https://doi.org/10.3390/ani11123548
Chicago/Turabian StyleCapozza, Paolo, Annamaria Pratelli, Michele Camero, Gianvito Lanave, Grazia Greco, Francesco Pellegrini, and Maria Tempesta. 2021. "Feline Coronavirus and Alpha-Herpesvirus Infections: Innate Immune Response and Immune Escape Mechanisms" Animals 11, no. 12: 3548. https://doi.org/10.3390/ani11123548
APA StyleCapozza, P., Pratelli, A., Camero, M., Lanave, G., Greco, G., Pellegrini, F., & Tempesta, M. (2021). Feline Coronavirus and Alpha-Herpesvirus Infections: Innate Immune Response and Immune Escape Mechanisms. Animals, 11(12), 3548. https://doi.org/10.3390/ani11123548