
Core Microbiome of Slovak Holstein Friesian Breeding Bulls’ Semen

 ,
,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Biological Material Sampling and Preparation
2.2. DNA Extraction and Illumina Library Preparation
2.3. Data Processing
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Somashekar, L.; Selvaraju, S.; Parthipan, S.; Ravindra, J.P. Profiling of sperm proteins and association of sperm PDC-109 with bull fertility. Syst. Biol. Reprod. Med. 2015, 61, 376–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthipan, S.; Selvaraju, S.; Somashekar, L.; Arangasamy, A.; Sivaram, M.; Ravindra, J.P. Spermatozoal transcripts expression levels are predictive of semen quality and conception rate in bulls (Bos taurus). Theriogenology 2017, 98, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Parthipan, S.; Somashekar, L.; Binsila, B.K.; Kolte, A.P.; Arangasamy, A.; Parameshwaraiah Ravindra, J.; Krawetz, S.A. Current status of sperm functional genomics and its diagnostic potential of fertility in bovine (Bos taurus). Syst. Biol. Reprod. Med. 2018, 64, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, I.M.; Dobson, H. Postpartum uterine health in cattle. Anim. Reprod. Sci. 2004, 82, 295–306. [Google Scholar] [CrossRef]
- Ashebir, G.; Birhanu, A.; Gugsa, T. Status of artificial insemination in tigray regional state, constraints and acceptability under field condition. J. Dair. Vet. Anim. Res. 2016, 3, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Sisay, T.A.; Amare, A.; Mekuriaw, Z. Quality evaluation of cryopreserved semen used in artificial insemination of cattle in selected districts of Western Gojjam zone of Amhara region, Ethiopia. J. Reprod. Infert. 2012, 3, 1–7. [Google Scholar]
- Sisay, W.; Tamene, D.; Worku, G.; Kidanu, D.; Getahun, B.; Nuraddis, I. Evaluation of artificial insemination efficiency in and around Ejere District, Western Shoa Zone, Ethiopia. J. Reprod. Infert. 2017, 8, 66–71. [Google Scholar] [CrossRef]
- Thibier, M.; Guerin, B. Hygienic aspects of storage and use of semen for artificial insemination. Anim. Reprod. Sci. 2000, 62, 233–251. [Google Scholar] [CrossRef]
- Yániz, J.L.; Marco-Aguado, M.A.; Mateos, J.A.; Santolaria, P. Bacterial contamination of ram semen, antibiotic sensitivities and effects on sperm quality during storage at 15 °C. Anim. Rep. Sci. 2010, 122, 142–149. [Google Scholar] [CrossRef]
- Bresciani, C.; Cabassi, C.S.; Morini, G.; Taddei, S.; Bettini, R.; Bigliardi, F.; Sabbioni, A.; Parmigiani, E. Boar semen bacterial contamination in Italy and antibiotic efficacy in a modified extender. It. J. Anim. Sci. 2014, 13, 3082. [Google Scholar] [CrossRef] [Green Version]
- Abro, S.H.; Abro, R.; Tunio, M.; Rind, R.; Bughio, S. Evidence of bacterial contamination in the frozen bovine Semen. Pak. J. Agricul. Agricul. Eng. Vet. Sci. 2015, 31, 102–108. [Google Scholar]
- Gączarzewicz, D.; Udała, J.; Piasecka, M.; Błaszczyk, B.; Stankiewicz, T. Bacterial contamination of boar semen and its relationship to sperm quality preserved in commercial extender containing gentamicin sulfate. Pol. J. Vet. Sci. 2016, 19, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Mitra, J.; Chowdhury, S.; Panda, S.; Chakraborty, M.; Singha, A. Microbiological evaluation of bovine frozen semen samples in West Bengal, India. Explor. Anim. Med. Res. 2016, 6, 185–191. [Google Scholar]
- González-Marin, C.; Roy, R.; López-Fernández, C.; Diez, B.; Carabaño, M.L.; Fernández, J.L.; Kjelland, M.E.; Moreno, J.F.; Gosálvez, J. Bacteria in bovine semen can increase sperm DNA fragmentation rates: A kinetic experimental approach. Anim. Rep. Sci. 2011, 123, 139–148. [Google Scholar] [CrossRef]
- Wierzbowski, S.; Nowakowski, W.; Wayda, E.; Kuzniak, S. Antibiotic level and bacterial contamination of frozen bull’s semen (streptomycin, penicillin). Med. Weteryn. 1984, 40, 284–287. [Google Scholar]
- Sannat, C.; Nair, A.; Sahu, S.B.; Sahasrabudhe, S.A.; Kumar, A.; Gupta, A.K.; Shende, R.K. Effect of species, breed, and age on bacterial load in bovine and bubaline semen. Vet. World 2015, 8, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Sannat, C.; Nair, A.; Sahu, S.B.; Sahasrabudhe, S.A.; Rawat, N.; Shende, R.K. Effect of season on bacterial load in semen of different breeds of cattle. J. Anim. Res. 2016, 6, 651–656. [Google Scholar] [CrossRef]
- Poiani, A. Complexity of seminal fluid: A review. Behav. Ecol. Sociobiol. 2006, 60, 289–310. [Google Scholar] [CrossRef]
- Schulze, M.; Jakop, U.; Schrooter, F.; Herrmann, C.; Leiding, C.; Müller, K.; Jung, M.; Czirják, G.A. Antibacterial defense in bull and boar semen: A putative link to the microbiome and reproductive strategy? Theriogenology 2020, 157, 335–340. [Google Scholar] [CrossRef]
- Reda, A.A.; Almaw, G.; Abreha, S.; Tadeg, W.; Tadesse, B. Bacteriospermia and Sperm Quality of Cryopreserved Bull Semen Used in Artificial Insemination of Cows in South Wollo Zone, Ethiopia. Vet. Med. Int. 2020, 2020, 2098315. [Google Scholar] [CrossRef] [Green Version]
- Monga, M.; Roberts, J.A. Spermagglutination by bacteria: Receptor-specific interactions. J. Androl. 1994, 15, 151–156. [Google Scholar]
- Köhn, F.M.; Erdmann, I.; Oeda, T.; el Mulla, K.F.; Schiefer, H.G.; Schill, W.B. Influence of urogenital infections on sperm functions. Andrologia. 1998, 30 (Suppl. S1), 73–80. [Google Scholar] [CrossRef]
- Althouse, G.C.; Pierdon, M.S.; Lu, K.G. Thermotemporal dynamics of contaminant bacteria and antimicrobials in extended porcine semen. Theriogenology 2008, 70, 1317–1323. [Google Scholar] [CrossRef]
- Bussalleu, E.; Yeste, M.; Sepúlveda, L.; Torner, E.; Pinart, E.; Bonet, S. Effects of different concentrations of enterotoxigenic and verotoxigenic E. coli on boar sperm quality. Anim. Reprod. Sci. 2011, 127, 176–182. [Google Scholar] [CrossRef]
- Ong, C.T.; Turni, C.; Blackall, P.J.; Boe-Hansen, G.; Hayes, B.J.; Tabor, A.E. Interrogating the bovine reproductive tract metagenomes using culture-independent approaches: A systematic review. Anim. Microbiom. 2021, 3, 41. [Google Scholar] [CrossRef]
- Ďuračka, M.; Kováčik, A.; Kačániová, M.; Lukáč, N.; Tvrdá, E. Bacteria may deteriorate progressive motility of bovine spermatozoa and biochemical parameters of seminal plasma. J. Microbiol. Biotechnol. Food Sci. 2020, 9, 844–847. [Google Scholar] [CrossRef]
- Risely, A. Applying the core microbiome to understand host–microbe systems. J. Anim. Ecol. 2020, 89, 1549–1558. [Google Scholar] [CrossRef] [Green Version]
- Björk, J.R.; O’Hara, R.B.; Ribes, M.; Coma, R.; Montoya, J.M. The Dynamic Core Microbiome: Structure, Dynamics and Stability. Available online: https://hal-univ-tlse3.archives-ouvertes.fr/hal-03043059 (accessed on 10 September 2021).
- Marchesi, J.R.; Ravel, J. The vocabulary of microbiome research: A proposal. Microbiome 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Sjöling, S.; Cowan, D.A. Metagenomics: Microbial community genomes revealed. In Psychrophiles: From Biodiversity to Biotechnology; Margesin, R., Schinner, F., Marx, J.C., Gerday, C., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 313–332. [Google Scholar]
- Shade, A.; Hogan, C.S.; Klimowicz, A.K.; Linske, M.; McManus, P.S.; Handelsman, J. Culturing captures members of the soil rare biosphere. Environ. Microbiol. 2012, 14, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Bragg, L.; Tyson, G.W. Metagenomics using next-generation sequencing. Methods Mol. Biol. 2014, 1096, 183–201. [Google Scholar] [CrossRef]
- Thomas, R.K.; Nickerson, E.; Simons, J.F.; Jänne, P.A.; Tengs, T.; Yuza, Y.; Garraway, L.A.; LaFramboise, T.; Lee, J.C.; Shah, K.; et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat. Med. 2006, 12, 852–855. [Google Scholar] [CrossRef]
- Heil, B.A.; Paccamonti, D.L.; Sones, J.L. Role for the mammalian female reproductive tract microbiome in pregnancy outcomes. Physiol. Genom. 2019, 51, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Wickware, C.L.; Johnson, T.A.; Koziol, J.H. Composition and diversity of the preputial microbiota in healthy bulls. Theriogenology 2020, 145, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Medo, J.; Žiarovská, J.; Medová, J.; Javoreková, S.; Kyseľ, M.; Hricová, A. Endophytic bacterial diversity decrease in amaranth mutant lines after radiation mutagenesis. Cereal Chem. 2018, 95, 109–116. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNAdiversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Větrovský, T.; Baldrian, P.; Morais, D. SEED 2: A user-friendly platform for amplicon high-throughput sequencing data analyses. Bioinformatics 2018, 34, 2292–2294. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Caporaso, J.G. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. [Google Scholar]
- Yang, H.; Zhang, J.; Xue, Z.; Zhao, C.H.; Lei, L.; Wen, Y.; Dong, Y.; Yang, J.; Zhang, L. Potential Pathogenic Bacteria in Seminal Microbiota of Patients with Different Types of Dysspermatism. Sci. Rep. 2020, 10, 6876. [Google Scholar] [CrossRef]
- Alfano, M.; Ferrarese, R.; Locatelli, I.; Ventimiglia, E.; Ippolito, S.; Gallina, P.; Cesana, D.; Canducci, F.; Pagliardini, L.; Viganò, P.; et al. Testicular microbiome in azoospermic men-first evidence of the impact of an altered microenvironment. Hum. Reprod. 2018, 33, 1212–1217. [Google Scholar] [CrossRef]
- Willeén, M.; Hoist, E.; Myhre, B.; Olsson, A.M. The bacterial flora of the genitourinary tract in healthy fertile men. Scand. J. Urol. Nephrol. 1996, 30, 387–393. [Google Scholar] [CrossRef]
- Givens, M. Review: Risks of disease transmission through semen in cattle. Animal 2018, 12, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Balzan, C.; Ziech, R.E.; Gressler, L.T.; de Vargas, A.P.C. Bovine genital campylobacteriosis: Main features and perspectives for diagnosis and control. Ciência Rural Santa Maria 2020, 50, e20190272. [Google Scholar] [CrossRef]
- Modolo, J.R.; Lopes, C.A.M.; Genari, T. Occurrence of Campylobacter in the genitals of teaser bulls maintained at an embryo transfer center. Braz. Arch. Vet. Med. Zootec. 2000, 52, 96–97. [Google Scholar] [CrossRef]
- Mshelia, G.D.; Amin, J.D.; Woldehiwet, Z.; Murray, R.D.; Egwu, G.O. Epidemiology of bovine venereal campylobacteriosis: Geographic distribution and recent advances in molecular diagnostic techniques. Reprod. Domest. Anim. 2010, 45, e221–e230. [Google Scholar] [CrossRef]
- Michi, A.N.; Favetto, P.H.; Kastelic, J.; Cobo, E.R. A review of sexually transmitted bovine trichomoniasis and campylobacteriosis affecting cattle reproductive health. Theriogenology 2016, 85, 781–791. [Google Scholar] [CrossRef]
- Baud, D.; Pattaroni, C.; Vulliemoz, N.; Castella, V.; Marsland, B.J.; Stojanov, M. Sperm microbiota and its impact on semen parameters. Front. Microbiol. 2019, 10, 234. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.L.; Chiu, C.M.; Lin, F.M.; Huang, W.C.; Liang, C.; Yang, T.; Yang, T.L.; Liu, C.Y.; Wu, W.Y.; Chang, Y.A.; et al. Bacterial communities in semen from men of infertile couples: Metagenomic sequencing reveals relationships of seminal microbiota to semen quality. PLoS ONE 2014, 9, e110152. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Meng, H.; Zhou, F.; Ni, X.; Shen, S.; Das, U.N. Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Archiv. Med. Sci. AMS 2015, 11, 385–394. [Google Scholar] [CrossRef]
- Ubeda, J.L.; Ausejo, R.; Dahmani, Y.; Falceto, M.V.; Usan, A.; Malo, C.; Perez-Martinez, F.C. Adverse effects of members of the Enterobacteriaceae family on boar sperm quality. Theriogenology 2013, 80, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.G. Achromobacter xylosoxidans in extended semen causes reproductive failure in artificially inseminated sows and gilts. J. Swine Health Prod. 2008, 16, 316–322. [Google Scholar]
- Rowe, M.; Veerus, L.; Trosvik, P.; Buckling, A.; Pizzari, T. The reproductive microbiome: An emerging driver of sexual selection, sexual conflict, mating systems, and reproductive isolation. Trends Ecol. Evol. 2020, 35, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.B. 16S rRNA gene sequencing for bacterial pathogen identification in the clinical laboratory. J. Mol. Diagn. 2001, 6, 313–321. [Google Scholar] [CrossRef]
- Ďuračka, M.; Belić, L.; Tokárová, K.; Žiarovská, J.; Kačániová, M.; Lukáč, N.; Tvrdá, E. Bacterial communities in bovine ejaculates and their impact on the semen quality. Syst. Biol. Reprod. Med. 2021, 27, 1–12. [Google Scholar] [CrossRef]
- Lenický, M.; Slanina, T.; Kačániová, M.; Galovičová, L.; Petrovičová, M.; Ďuračka, M.; Benko, F.; Kováč, J.; Tvrdá, E. Identification of Bacterial Profiles and Their Interactions with Selected Quality, Oxidative, and Immunological Parameters of Turkey Semen. Animals 2021, 11, 1771. [Google Scholar] [CrossRef]
- Patwardhan, A.; Ray, S.; Roy, A. Molecular markers in phylogenetic studies-a review. J. Phylogen. Evol. Biol. 2014, 2, 1000131. [Google Scholar] [CrossRef]
- Al-Kass, Z.; Eriksson, E.; Bagge, E.; Wallgren, M.; Morrell, J. Microbiota of semen from stallions in Sweden identified by MALDI-TOF. Vet. Anim. Sci. 2020, 10, 100143. [Google Scholar] [CrossRef]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [Green Version]

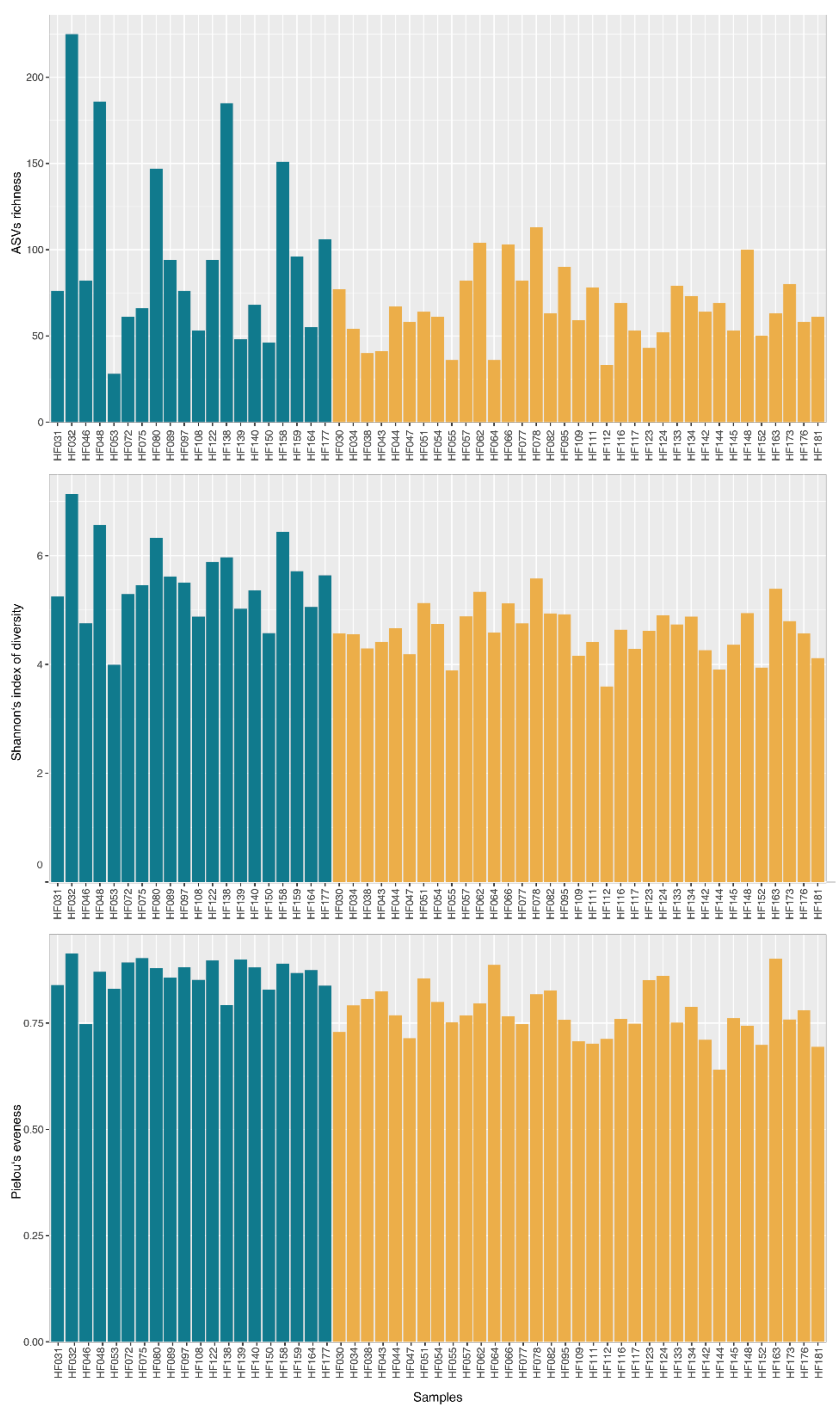
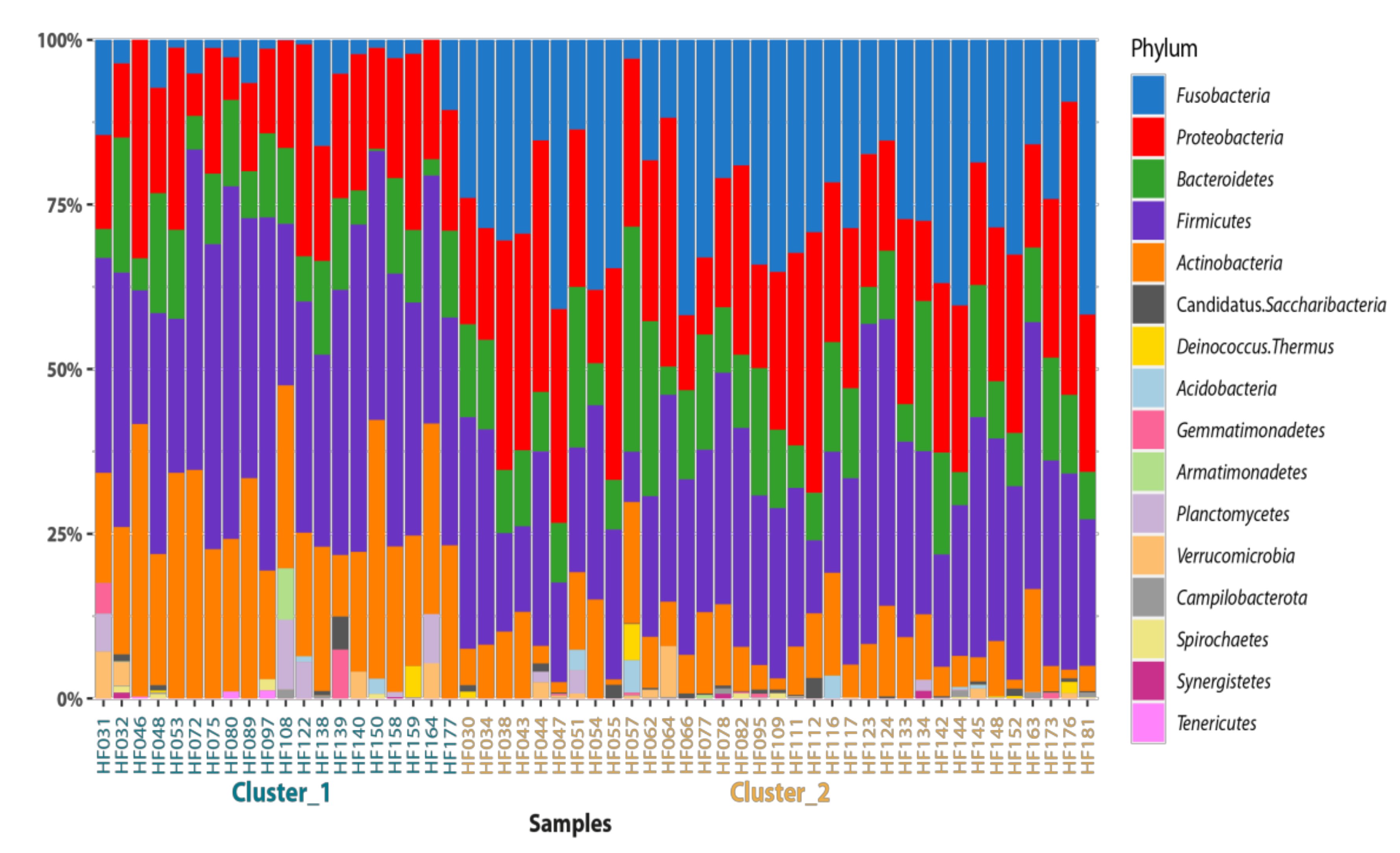

{kind=link}
{kind=link}
{kind=link}
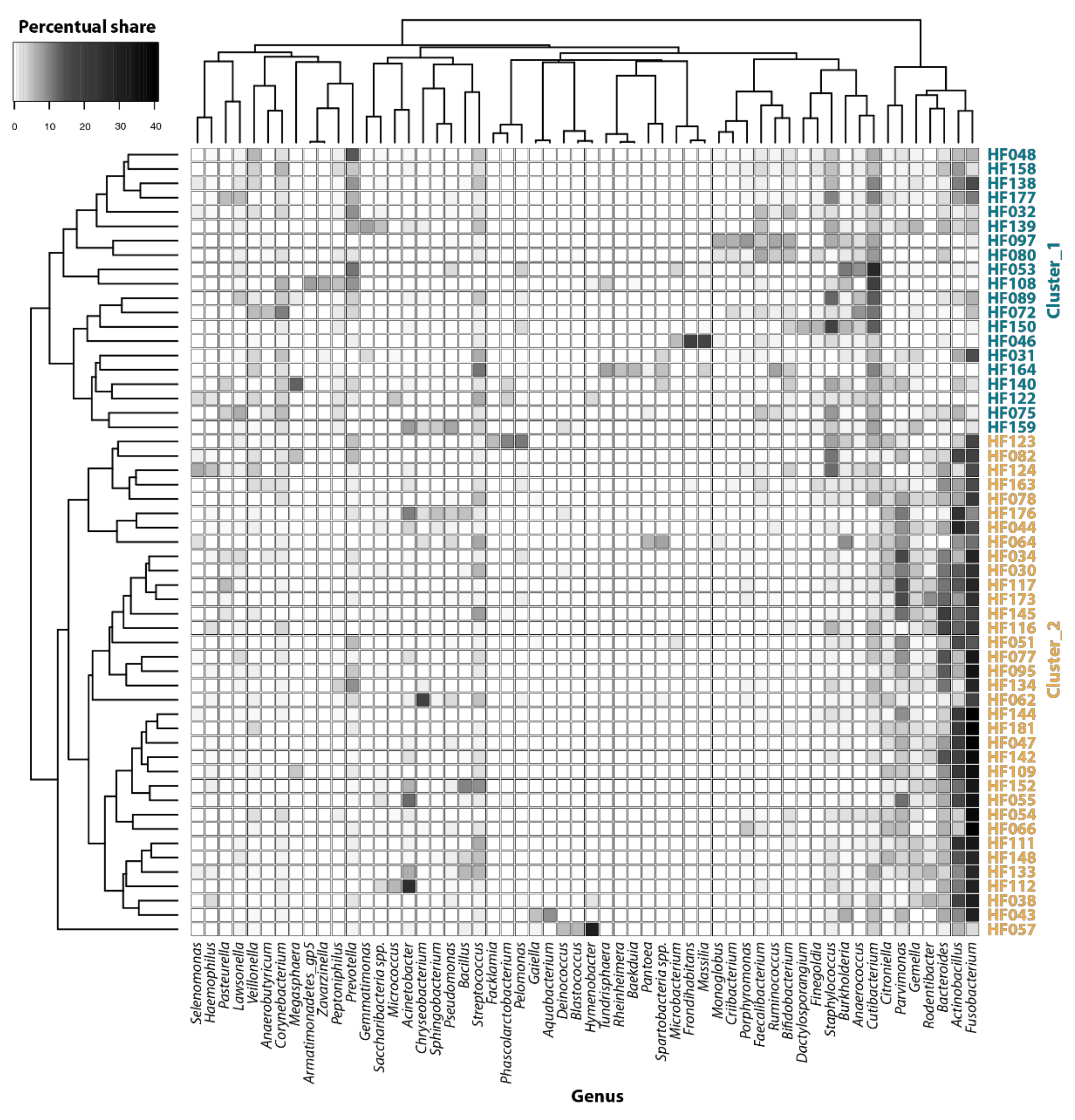
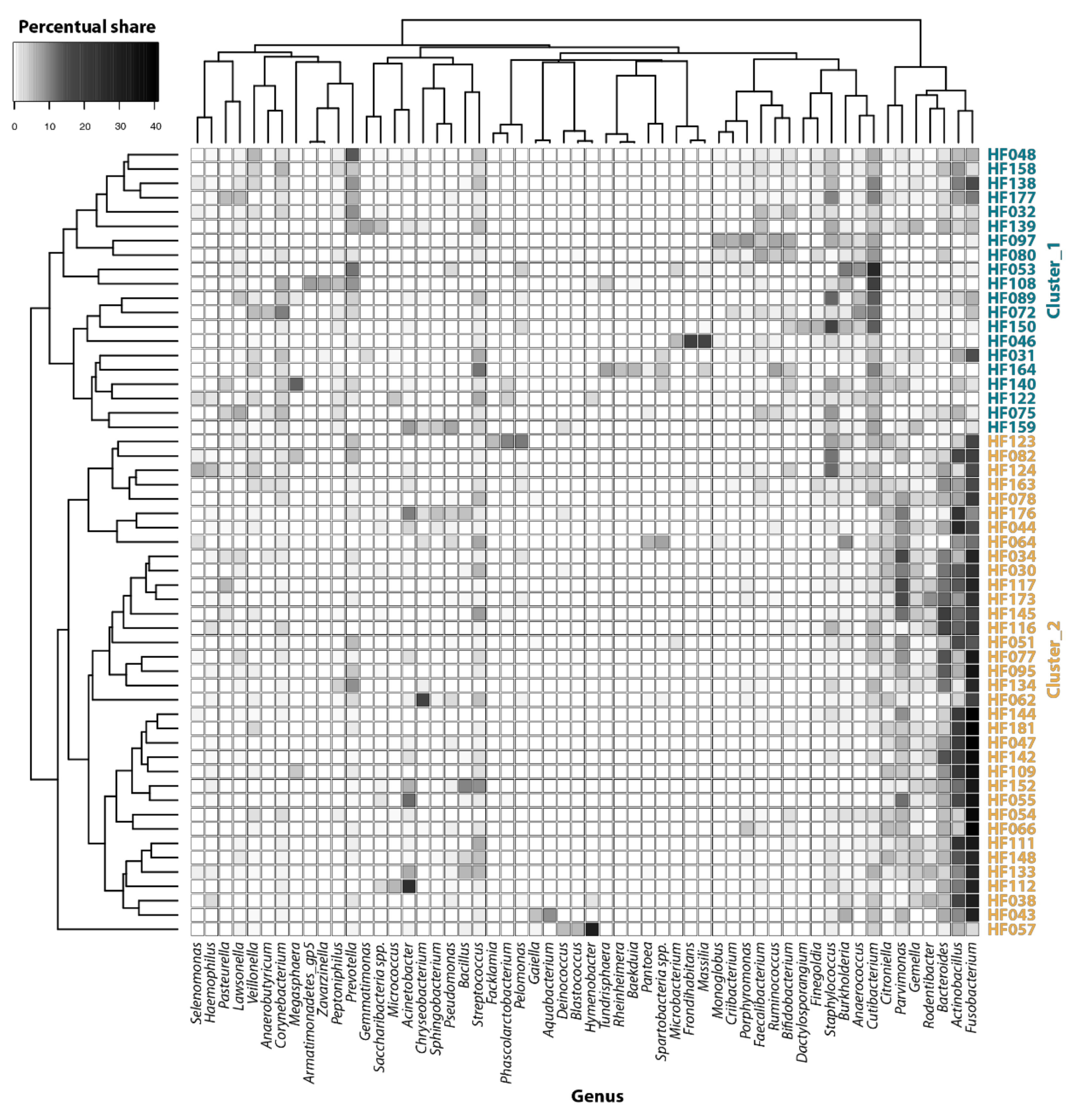{kind=link}
| Phylum | Class | Order | Family | Genus | ASV | Average Share ± SE in Cluster 1 | Number of Samples Detected in Cluster 1 (n = 20) | Average Share ± SE in Cluster 2 | Number of Samples Detected in Cluster 2 (n = 35) | p Value |
|---|---|---|---|---|---|---|---|---|---|---|
| Actinobacteria | 24.57 ± 1.87 | 20 | 7.74 ± 0.8 | 35 | 0.001 | |||||
| Actinobacteria | 23.55 ± 1.94 | 20 | 7.33 ± 0.74 | 35 | 0.001 | |||||
| Bifidobacteriales | 2.18 ± 0.45 | 17 | 0.53 ± 0.14 | 23 | 0.001 | |||||
| Bifidobacteriaceae | 2.18 ± 0.45 | 17 | 0.53 ± 0.14 | 23 | 0.001 | |||||
| Bifidobacterium | 2.17 ± 0.45 | 17 | 0.51 ± 0.14 | 23 | 0.001 | |||||
| Micrococcales | 3.76 ± 1.67 | 17 | 0.87 ± 0.19 | 32 | 0.003 | |||||
| Microbacteriaceae | 2.16 ± 1.59 | 13 | 0.19 ± 0.07 | 14 | 0.022 | |||||
| Mycobacteriales | 5.39 ± 0.85 | 20 | 1.78 ± 0.23 | 34 | 0.001 | |||||
| Corynebacteriaceae | 3.37 ± 0.56 | 18 | 1.13 ± 0.16 | 33 | 0.001 | |||||
| Corynebacterium | 3.37 ± 0.56 | 18 | 1.13 ± 0.16 | 33 | 0.001 | |||||
| Propionibacteriales | 9.88 ± 1.3 | 20 | 3.11 ± 0.35 | 35 | 0.001 | |||||
| Propionibacteriaceae | 9.31 ± 1.3 | 20 | 2.99 ± 0.35 | 35 | 0.001 | |||||
| Cutibacterium | 9.15 ± 1.28 | 20 | 2.85 ± 0.33 | 35 | 0.001 | |||||
| ASV7 | 7.92 ± 1.03 | 20 | 2.69 ± 0.32 | 35 | 0.001 | |||||
| Bacteroidetes | 10.17 ± 1.2 | 20 | 12.73 ± 1.14 | 35 | 0.267 | |||||
| Bacteroidia | 8.73 ± 1.26 | 20 | 10.12 ± 0.86 | 34 | 0.319 | |||||
| Bacteroidales | 8.73 ± 1.26 | 20 | 10.12 ± 0.86 | 34 | 0.319 | |||||
| Bacteroidaceae | 2.26 ± 0.53 | 16 | 7.65 ± 0.78 | 34 | 0.001 | |||||
| Bacteroides | 1.68 ± 0.42 | 14 | 7.42 ± 0.79 | 34 | 0.001 | |||||
| ASV4 | 0.82 ± 0.33 | 9 | 6.47 ± 0.68 | 34 | 0.001 | |||||
| Prevotellaceae | 4.83 ± 0.99 | 18 | 1.81 ± 0.36 | 28 | 0.015 | |||||
| Prevotella | 4.31 ± 0.94 | 18 | 1.64 ± 0.34 | 28 | 0.028 | |||||
| Firmicutes | 38.08 ± 2.1 | 20 | 26.63 ± 1.52 | 35 | 0.001 | |||||
| Bacilli | 12.18 ± 1.12 | 20 | 9.02 ± 0.87 | 35 | 0.027 | |||||
| Bacillales | 7.06 ± 1.06 | 20 | 5.47 ± 0.58 | 35 | 0.228 | |||||
| Bacillales_Incertae Sedis XI | 1.28 ± 0.38 | 14 | 2.23 ± 0.24 | 34 | 0.005 | |||||
| Gemella | 1.28 ± 0.38 | 14 | 2.23 ± 0.24 | 34 | 0.005 | |||||
| Staphylococcaceae | 5.19 ± 1.01 | 19 | 2.28 ± 0.48 | 34 | 0.005 | |||||
| Staphylococcus | 5.18 ± 1.01 | 19 | 2.27 ± 0.48 | 34 | 0.005 | |||||
| ASV9 | 4.51 ± 0.95 | 19 | 1.96 ± 0.44 | 33 | 0.003 | |||||
| Lactobacillales | 5.12 ± 0.58 | 20 | 3.54 ± 0.49 | 35 | 0.025 | |||||
| Streptococcaceae | 4.07 ± 0.56 | 20 | 2.86 ± 0.41 | 34 | 0.033 | |||||
| Streptococcus | 3.33 ± 0.62 | 19 | 2.72 ± 0.41 | 34 | 0.354 | |||||
| Clostridia | 22.25 ± 2.35 | 20 | 15.24 ± 1.1 | 35 | 0.039 | |||||
| Clostridiales | 22.25 ± 2.35 | 20 | 15.24 ± 1.1 | 35 | 0.039 | |||||
| Lachnospiraceae | 6.08 ± 0.96 | 20 | 1.52 ± 0.3 | 32 | 0.001 | |||||
| Peptoniphilaceae | 7.45 ± 0.7 | 20 | 12.13 ± 0.92 | 35 | 0.002 | |||||
| Anaerococcus | 2.27 ± 0.57 | 17 | 0.51 ± 0.12 | 28 | 0.001 | |||||
| Citroniella | 0.64 ± 0.25 | 11 | 2.63 ± 0.32 | 33 | 0.001 | |||||
| ASV11 | 0.46 ± 0.22 | 9 | 2.58 ± 0.31 | 33 | 0.001 | |||||
| Helcococcus | 0.39 ± 0.15 | 12 | 2 ± 0.21 | 33 | 0.001 | |||||
| Parvimonas | 0.99 ± 0.33 | 16 | 6.15 ± 0.76 | 35 | 0.001 | |||||
| ASV5 | 0.99 ± 0.33 | 16 | 6.15 ± 0.76 | 35 | 0.001 | |||||
| Ruminococcaceae | 6.65 ± 1.45 | 19 | 0.95 ± 0.18 | 32 | 0.001 | |||||
| Faecalibacterium | 2.25 ± 0.48 | 18 | 0.4 ± 0.12 | 22 | 0.001 | |||||
| Negativicutes | 3.52 ± 0.93 | 15 | 2.19 ± 0.52 | 33 | 0.468 | |||||
| Veillonellales | 2.75 ± 0.69 | 14 | 1.34 ± 0.31 | 31 | 0.204 | |||||
| Veillonellaceae | 2.75 ± 0.69 | 14 | 1.34 ± 0.31 | 31 | 0.204 | |||||
| Fusobacteria | 4.23 ± 1.05 | 18 | 26.28 ± 1.69 | 35 | 0.001 | |||||
| Fusobacteriia | 4.23 ± 1.05 | 18 | 26.28 ± 1.69 | 35 | 0.001 | |||||
| Fusobacteriales | 4.23 ± 1.05 | 18 | 26.28 ± 1.69 | 35 | 0.001 | |||||
| Fusobacteriaceae | 3.96 ± 1.05 | 18 | 26.13 ± 1.68 | 35 | 0.001 | |||||
| Fusobacterium | 3.96 ± 1.05 | 18 | 26.13 ± 1.68 | 35 | 0.001 | |||||
| ASV1 | 1.73 ± 0.78 | 10 | 9.68 ± 1.23 | 34 | 0.001 | |||||
| ASV2 | 0.13 ± 0.08 | 4 | 3.16 ± 0.53 | 29 | 0.001 | |||||
| ASV8 | 0.91 ± 0.38 | 10 | 9.41 ± 1.28 | 34 | 0.001 | |||||
| ASV10 | 0.75 ± 0.33 | 7 | 3.69 ± 0.47 | 32 | 0.001 | |||||
| Proteobacteria | 18.14 ± 1.62 | 20 | 24.67 ± 1.42 | 35 | 0.007 | |||||
| Alphaproteobacteria | 2.3 ± 0.66 | 19 | 1.3 ± 0.46 | 30 | 0.041 | |||||
| Betaproteobacteria | 6.69 ± 1.36 | 20 | 3.54 ± 0.73 | 34 | 0.005 | |||||
| Burkholderiales | 5.43 ± 1.37 | 20 | 2.79 ± 0.71 | 34 | 0.016 | |||||
| Burkholderiaceae | 2.64 ± 0.77 | 20 | 1.54 ± 0.33 | 34 | 0.064 | |||||
| Burkholderia | 2.22 ± 0.6 | 20 | 1.47 ± 0.32 | 34 | 0.298 | |||||
| ASV17 | 2.22 ± 0.6 | 20 | 1.45 ± 0.32 | 34 | 0.278 | |||||
| Gammaproteobacteria | 8.87 ± 1.46 | 20 | 19.72 ± 1.57 | 35 | 0.001 | |||||
| Pasteurellales | 5.23 ± 1.12 | 19 | 15.45 ± 1.29 | 35 | 0.001 | |||||
| Pasteurellaceae | 5.23 ± 1.12 | 19 | 15.45 ± 1.29 | 35 | 0.001 | |||||
| Actinobacillus | 2.99 ± 0.76 | 17 | 12.05 ± 1.17 | 35 | 0.001 | |||||
| ASV3 | 1.7 ± 0.43 | 14 | 6.31 ± 0.69 | 34 | 0.001 | |||||
| ASV6 | 1.29 ± 0.39 | 13 | 5.73 ± 0.6 | 34 | 0.001 | |||||
| Rodentibacter | 0.25 ± 0.13 | 6 | 2.11 ± 0.35 | 30 | 0.001 | |||||
| Pseudomonadales | 1.87 ± 0.76 | 16 | 3.25 ± 0.88 | 33 | 0.214 | |||||
| Moraxellaceae | 1.25 ± 0.48 | 14 | 2.6 ± 0.83 | 31 | 0.251 | |||||
| Acinetobacter | 0.76 ± 0.39 | 10 | 2.36 ± 0.82 | 25 | 0.089 | |||||
| ASV13 | 0.63 ± 0.4 | 7 | 2.21 ± 0.83 | 20 | 0.086 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medo, J.; Žiarovská, J.; Ďuračka, M.; Tvrdá, E.; Baňas, Š.; Gábor, M.; Kyseľ, M.; Kačániová, M. Core Microbiome of Slovak Holstein Friesian Breeding Bulls’ Semen. Animals 2021, 11, 3331. https://doi.org/10.3390/ani11113331
Medo J, Žiarovská J, Ďuračka M, Tvrdá E, Baňas Š, Gábor M, Kyseľ M, Kačániová M. Core Microbiome of Slovak Holstein Friesian Breeding Bulls’ Semen. Animals. 2021; 11(11):3331. https://doi.org/10.3390/ani11113331
Chicago/Turabian StyleMedo, Juraj, Jana Žiarovská, Michal Ďuračka, Eva Tvrdá, Štefan Baňas, Michal Gábor, Matúš Kyseľ, and Miroslava Kačániová. 2021. "Core Microbiome of Slovak Holstein Friesian Breeding Bulls’ Semen" Animals 11, no. 11: 3331. https://doi.org/10.3390/ani11113331
APA StyleMedo, J., Žiarovská, J., Ďuračka, M., Tvrdá, E., Baňas, Š., Gábor, M., Kyseľ, M., & Kačániová, M. (2021). Core Microbiome of Slovak Holstein Friesian Breeding Bulls’ Semen. Animals, 11(11), 3331. https://doi.org/10.3390/ani11113331