
ssGBLUP Method Improves the Accuracy of Breeding Value Prediction in Huacaya Alpaca


 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutiérrez, J.P.; Goyache, F.; Burgos, A.; Cervantes, I. Genetic analysis of six production traits in Peruvian alpacas. Livest. Sci. 2009, 123, 193–197. [Google Scholar] [CrossRef]
- Cruz, A.; Morante, R.; Gutiérrez, J.P.; Torres, R.; Burgos, A.; Cervantes, I. Genetic parameters for medullated fiber and its relationship with other productive traits in alpacas. Animal 2019, 13, 1358–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, J.P.; Varona, L.; Pun, A.; Morante, R.; Burgos, A.; Cervantes, I.; Perez-Cabal, M.A. Genetic parameters for growth of fiber diameter in alpacas. J. Anim. Sci. 2011, 89, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.; Morante, R.; Cervantes, I.; Burgos, A.; Gutiérrez, J.P. Effect of the gestation and lactation on fiber diameter and its variability in Peruvian alpacas. Livest. Sci. 2017, 198, 31–36. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Legarra, A.; Aguilar, I.; Misztal, I. A relationship matrix including full pedigree and genomic information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, O.F.; Lund, M.S. Genomic prediction when some animals are not genotyped. Genet. Sel. Evol. 2010, 42, 2. [Google Scholar] [CrossRef] [Green Version]
- Legarra, A.; Christensen, O.F.; Aguilar, I.; Misztal, I. Single Step, a general approach for genomic selection. Livest. Sci. 2014, 166, 54–65. [Google Scholar] [CrossRef]
- Rupp, R.; Mucha, S.; Larroque, H.; McEwan, J.; Conington, J. Genomic application in sheep and goat breeding. Anim. Front. 2016, 6, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Teng, J.; Pan, R.; Li, X.; Ye, S.; Li, J.; Zhang, H.; Zhang, X.; Zhang, Z. Accuracy of whole genome prediction with single-step GBLUP in a Chinese yellow-feathered chicken population. Livest. Sci. 2019, 230. [Google Scholar] [CrossRef]
- Song, H.; Zhang, J.; Zhang, Q.; Ding, X. Using Different Single-Step Strategies to Improve the Efficiency of Genomic Prediction on Body Measurement Traits in Pig. Front. Genet. 2018, 9, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Chen, C.Y.; Misztal, I.; Aguilar, I.; Tsuruta, S.; Meuwissen, T.H.; Aggrey, S.E.; Wing, T.; Muir, W.M. Genome-wide marker-assisted selection combining all pedigree phenotypic information with genotypic data in one step: An example using broiler chickens. J. Anim. Sci. 2011, 89, 23–28. [Google Scholar] [CrossRef] [Green Version]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.; Hayes, B.; Goddard, M. Accelerating improvement of livestock with genomic selection. Annu. Rev. Anim. Biosci. 2013, 1, 221–237. [Google Scholar] [CrossRef]
- Calderon, M.; More, M.J.; Gutiérrez, G.A.; Ponce de León, F.A. Development of a 76k Alpaca (Vicugna pacos) Single Nucleotide Polymorphisms (SNPs) Microarray. Genes 2021, 12, 291. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, L.R.; Wilton, J.W.; Thompson, R. Simultaneous Estimation of Variance and Covariance Components from Multitrait Mixed Model Equations. Biometrics 1978, 34, 199–208. [Google Scholar] [CrossRef]
- Meyer, K. Maximum Likelihood Estimation of Variance Components for a Multivariate Mixed Model with Equal Design Matrices. Biometrics 1985, 41, 153–165. [Google Scholar] [CrossRef]
- Cruz, A. Parámetros Genéticos de Caracteres Funcionales y Secundarios en Alpacas. Doctoral Thesis, Universidad Complutense de Madrid, Madrid, Spain, 2017. [Google Scholar]
- Misztal, I.; Legarra, A.; Aguilar, I. Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J. Dairy Sci. 2009, 92, 4648–4655. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Misztal, I.; Lourenco, D.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs; University of Georgia: Athens, GA, USA, 2015. [Google Scholar]
- Quispe, E.C.; Rodríguez, T.C.; Iñiguez, L.R.; Mueller, J.P. Producción de fibra de alpaca, llama, vicuña y guanaco en Sudamérica. Anim. Genet. Resour. 2009, 45, 1–14. [Google Scholar] [CrossRef]
- Silva, M.V.B.; dos Santos, D.J.A.; Boison, S.A.; Utsunomiya, A.T.H.; Carmo, A.S.; Sonstegard, T.S.; Cole, J.B.; Van Tassell, C.P. The development of genomics applied to dairy breeding. Livest. Sci. 2014, 166, 66–75. [Google Scholar] [CrossRef]
- Pérez-Cabal, M.A.; Cervantes, I.; Morante, R.; Burgos, A.; Goyache, F.; Gutierrez, J.P. Analysis of the existence of major genes affecting alpaca fiber traits. J. Anim. Sci. 2010, 88, 3783–3788. [Google Scholar] [CrossRef] [Green Version]
- Mamani, C.; Gutiérrez, G.; Ponce de León, F.A. Identificación de polimorfismos de nucleótido simple en alpaca (Vicugna pacos) usando un panel de células híbridas irradiadas alpaca/hámster. Rev. Investig. Cienc. Y Biotecnol. Anim. 2017, 1, 92–95. [Google Scholar] [CrossRef]
- Mendoza, M.N.; Raudsepp, T.; Alshanbari, F.; Gutierrez, G.; Ponce de Leon, F.A. Chromosomal Localization of Candidate Genes for Fiber Growth and Color in Alpaca (Vicugna pacos). Front. Genet. 2019, 10, 583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, J.; Li, Q.; Wang, Q.; Wen, J.; Zhao, G. Comparison of the Efficiency of BLUP and GBLUP in Genomic Prediction of Immune Traits in Chickens. Animals 2020, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, M.; Mullaart, E.; Van der Linde, C.; Mulder, H.A. Estimating variance components and breeding values for number of oocytes and number of embryos in dairy cattle using a single-step genomic evaluation. J. Dairy Sci. 2017, 100, 4698–4705. [Google Scholar] [CrossRef] [Green Version]
- Misztal, I.; Aggrey, S.E.; Muir, W.M. Experiences with a single-step genome evaluation. Poult. Sci. 2013, 92, 2530–2534. [Google Scholar] [CrossRef]
- Akanno, E.C.; Schenkel, F.S.; Sargolzaei, M.; Friendship, R.M.; Robinson, J.A. Persistency of accuracy of genomic breeding values for different simulated pig breeding programs in developing countries. J. Anim. Breed. Genet. 2014, 131, 367–378. [Google Scholar] [CrossRef]
- Meuwissen, T.; Hayes, B.; Goddard, M. Genomic selection: A paradigm shift in animal breeding. Anim. Front. 2016, 6, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Swan, A.A.; van der Werf, J.H.; Hayes, B.J. Accuracy of pedigree and genomic predictions of carcass and novel meat quality traits in multi-breed sheep data assessed by cross-validation. Genet. Sel. Evol. 2012, 44, 33. [Google Scholar] [CrossRef] [Green Version]
- Legarra, A.; Baloche, G.; Barillet, F.; Astruc, J.M.; Soulas, C.; Aguerre, X.; Arrese, F.; Mintegi, L.; Lasarte, M.; Maeztu, F.; et al. Within- and across-breed genomic predictions and genomic relationships for Western Pyrenees dairy sheep breeds Latxa, Manech, and Basco-Bearnaise. J. Dairy Sci. 2014, 97, 3200–3212. [Google Scholar] [CrossRef]
- Teissier, M.; Larroque, H.; Robert-Granie, C. Weighted single-step genomic BLUP improves accuracy of genomic breeding values for protein content in French dairy goats: A quantitative trait influenced by a major gene. Genet. Sel. Evol. 2018, 50, 31. [Google Scholar] [CrossRef]
- Muir, W.M. Comparison of genomic and traditional BLUP-estimated breeding value accuracy and selection response under alternative trait and genomic parameters. J. Anim. Breed. Genet. 2007, 124, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Bowman, P.J.; Chamberlain, A.J.; Goddard, M.E. Invited review: Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci. 2009, 92, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, B.J.; Visscher, P.M.; Goddard, M.E. Increased accuracy of artificial selection by using the realized relationship matrix. Genet. Res. 2009, 91, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Daetwyler, H.D.; Hickey, J.M.; Henshall, J.M.; Dominik, S.; Gredler, B.; van der Werf, J.H.J.; Hayes, B.J. Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population. Anim. Prod. Sci. 2010, 50. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, D.A.; Tsuruta, S.; Fragomeni, B.O.; Masuda, Y.; Aguilar, I.; Legarra, A.; Bertrand, J.K.; Amen, T.S.; Wang, L.; Moser, D.W.; et al. Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. J. Anim. Sci. 2015, 93, 2653–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baby, S.; Hyeong, K.E.; Lee, Y.M.; Jung, J.H.; Oh, D.Y.; Nam, K.C.; Kim, T.H.; Lee, H.K.; Kim, J.J. Evaluation of genome based estimated breeding values for meat quality in a berkshire population using high density single nucleotide polymorphism chips. Asian-Australas. J. Anim. Sci. 2014, 27, 1540–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Erbe, M.; He, J.; Ober, U.; Gao, N.; Zhang, H.; Simianer, H.; Li, J. Accuracy of whole-genome prediction using a genetic architecture-enhanced variance-covariance matrix. Genes Genomes Genet. 2015, 5, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Cruz, A.; Cervantes, I.; Burgos, A.; Morante, R.; Gutierrez, J.P. Estimation of genetic parameters for reproductive traits in alpacas. Anim. Reprod. Sci. 2015, 163, 48–55. [Google Scholar] [CrossRef]


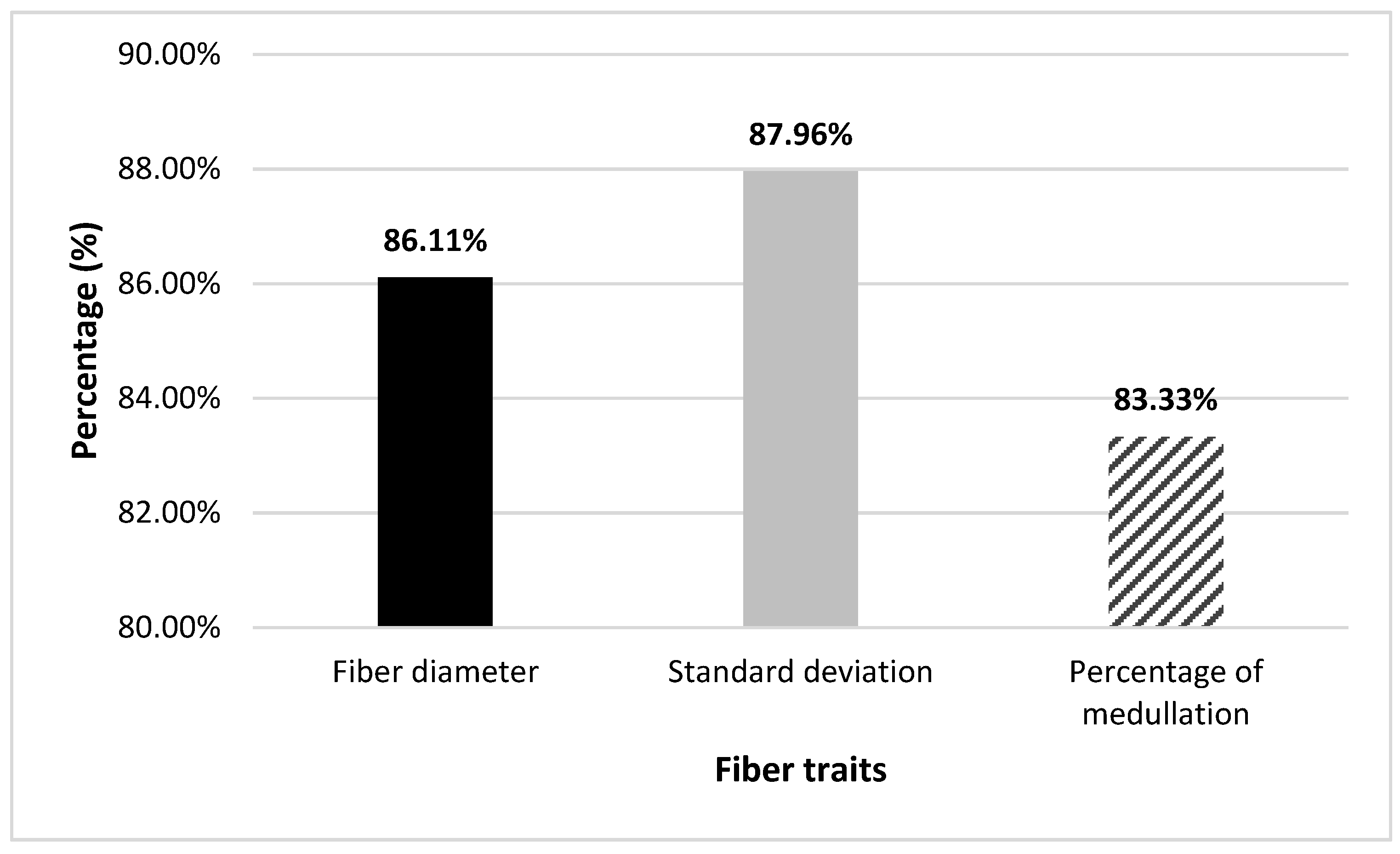{kind=link}
| Animals (n) | Total Records | |||
|---|---|---|---|---|
| FD | SD | PM | ||
| Full pedigree | 12,431 | |||
| Animal with records | 6889 | 24,169 | 24,169 | 8386 |
| Genotyped animals | 431 | 2774 | 2774 | 1767 |
| Methodology | Traits | h2 | |||
| BLUP | FD | 2.824 *** ± 0.002 | 1.289 *** ± 0.002 | 4.332 ns ± 0.001 | 0.334 ns ± 0.001 |
| SD | 0.354 ns ± 0.001 | 0.144 ns ± 0.001 | 0.431 ns ± 0.001 | 0.381 ns ± 0.001 | |
| PM | 27.416 *** ± 0.089 | 39.509 *** ±0.079 | 106.316 ns ± 0.128 | 0.158 ns ± 0.001 | |
| ssGBLUP | FD | 2.842 *** ± 0.002 | 1.280 *** ± 0.002 | 4.331 ns ± 0.001 | 0.336 ns ± 0.001 |
| SD | 0.355 ns ± 0.001 | 0.143 ns ± 0.001 | 0.431 ns ± 0.001 | 0.382 ns ± 0.001 | |
| PM | 27.717 *** ± 0.089 | 38.965 *** ± 0.077 | 106.352 ns ± 0.128 | 0.160 ns ± 0.001 |
| Traits | BLUP 1 | SsGBLUP 2 | Difference | Increase (%) |
|---|---|---|---|---|
| FD | 0.505 ± 0.015 | 0.517 ± 0.011 | 0.012 * ± 0.003 | 2.623 |
| SD | 0.445 ± 0.019 | 0.472 ± 0.015 | 0.027 ** ± 0.004 | 6.442 |
| PM | 0.308 ± 0.017 | 0.311 ± 0.013 | 0.004 ns ± 0.003 | 1.471 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancisidor, B.; Cruz, A.; Gutiérrez, G.; Burgos, A.; Morón, J.A.; Wurzinger, M.; Gutiérrez, J.P. ssGBLUP Method Improves the Accuracy of Breeding Value Prediction in Huacaya Alpaca. Animals 2021, 11, 3052. https://doi.org/10.3390/ani11113052
Mancisidor B, Cruz A, Gutiérrez G, Burgos A, Morón JA, Wurzinger M, Gutiérrez JP. ssGBLUP Method Improves the Accuracy of Breeding Value Prediction in Huacaya Alpaca. Animals. 2021; 11(11):3052. https://doi.org/10.3390/ani11113052
Chicago/Turabian StyleMancisidor, Betsy, Alan Cruz, Gustavo Gutiérrez, Alonso Burgos, Jonathan Alejandro Morón, Maria Wurzinger, and Juan Pablo Gutiérrez. 2021. "ssGBLUP Method Improves the Accuracy of Breeding Value Prediction in Huacaya Alpaca" Animals 11, no. 11: 3052. https://doi.org/10.3390/ani11113052