
The Quest for Genes Involved in Adaptation to Climate Change in Ruminant Livestock

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
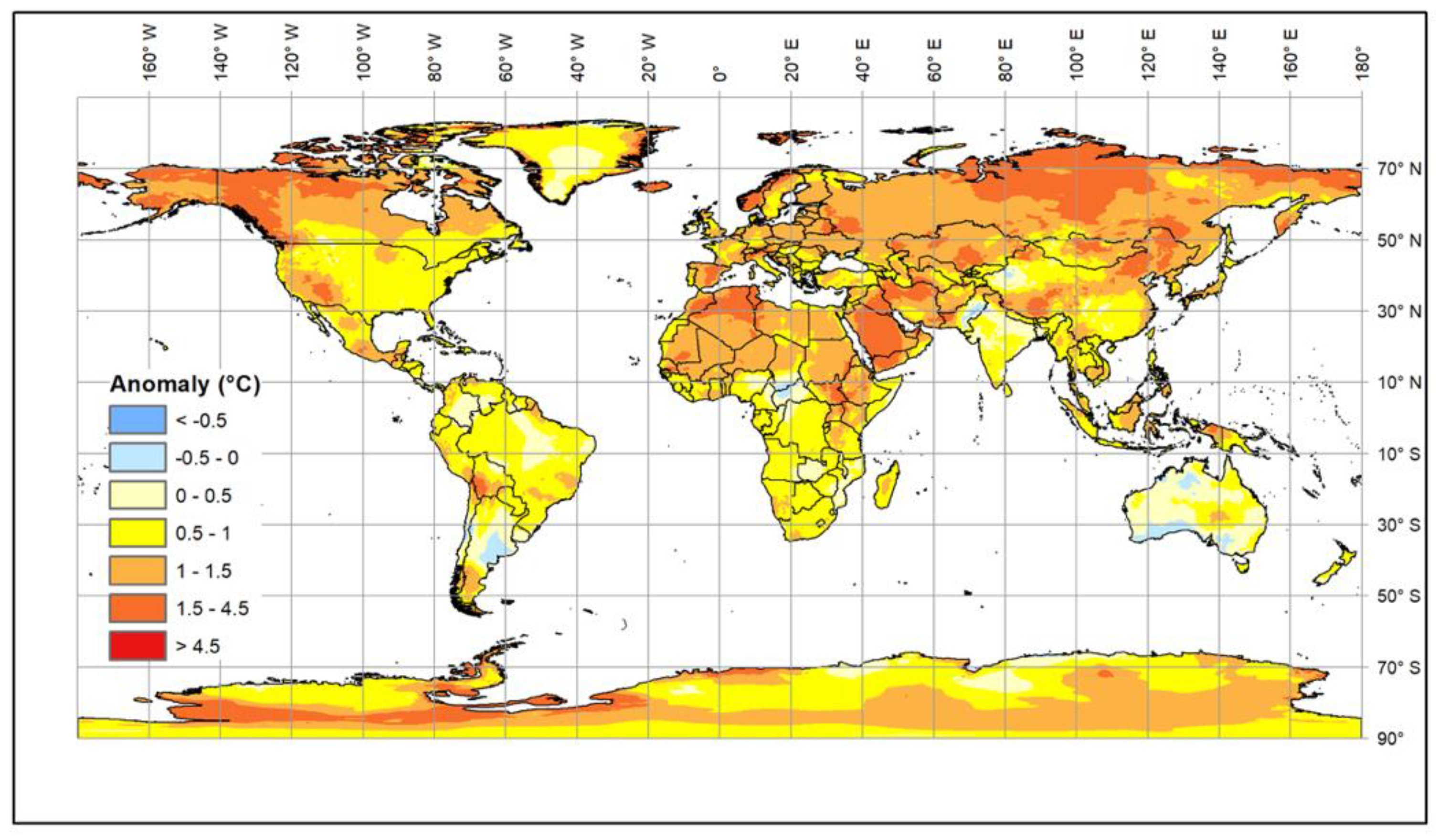
2. Impacts of Climate Change on Livestock
3. Becoming Adapted
4. Seeking Adaptive Genes
4.1. Genome-Wide Association Studies
4.2. Selection Signatures
4.3. Local Ancestry Inference
4.4. Landscape Genomics
4.5. Artificial Intelligence and Machine Learning Approaches

{kind=link}
| Software | Method | Application | Ref. | Link |
|---|---|---|---|---|
| Arlequin | Tajima’s D | Selection signatures | [254] | http://cmpg.unibe.ch/software/arlequin35/ |
| BayeScan | FST | Selection Signatures, Landscape genomics | [229] | http://cmpg.unibe.ch/software/BayeScan/ |
| Bcftools | ROH | Selection signatures | [255] | https://github.com/samtools/bcftools |
| DnaSP | Tajima’s D and Fay and Wu’s statistic | Selection signatures | http://www.ub.edu/dnasp/ | |
| Hapbin | EHH | Selection signatures | [256] | https://github.com/evotools/hapbin |
| hapFLK | hapFLK | Selection signatures | [179] | https://forge-dga.jouy.inra.fr/projects/hapflk |
| HierFstat (R package) | FST | Selection signatures | [257] | https://cran.r-project.org/web/packages/hierfstat/index.html |
| KING | ROH | Selection signatures | [258] | https://www.kingrelatedness.com/ |
| PLINK | FST, ROH | GWAS, Selection Signatures | [259] | https://www.cog-genomics.org/plink/2.0/https://www.cog-genomics.org/plink/ |
| PopGenome | Tajima’s D | Selection signatures | [260] | https://cran.r-project.org/web/packages/PopGenome/index.html |
| PoPoolation | Tajima’s D | Selection signatures | [261] | https://sourceforge.net/p/popoolation/wiki/Main/ |
| rehh (R package) | EHH | Selection signatures | [262] | https://cran.r-project.org/web/packages/rehh/index.html |
| Selscan | EHH | Selection signatures | [263] | https://github.com/szpiech/selscan |
| VariScan | Tajima’s D | Selection signatures | [264] | http://www.ub.edu/softevol/variscan/ |
| VCFtools | FST, Tajima’s D | Selection signatures | [265] | http://vcftools.sourceforge.net/ |
| EMMAX | GWAS based on variance component model | GWAS | [266] | http://genetics.cs.ucla.edu/emmax |
| GCTA | GWAS based on genome-wide complex trait analysis | GWAS | [267] | http://gump.qimr.edu.au/gcta |
| BayesR | Bayesian mixture model | GWAS | [268] | http://www.cnsgenomics.com/software/ |
| MatSAM | Logistic regression | Landscape genomics | [208] | www.econogene.eu/software/sam/ |
| Samβada, R.SamBada (R package) | GEA based on logistic regression/spatial autocorrelation | Landscape genomics | [213,219] | https://github.com/Sylvie/sambada/releases/tag/v0.8.3https://cran.r-project.org/package=R.SamBada |
| BAYENV | GEA based on Bayesian regression | Landscape genomics | [220] | https://gcbias.org/bayenv/ |
| LFMM2 (R package) | GEA based on latent factor mixed models | Landscape genomics | [221,226] | https://bcm-uga.github.io/lfmm/ |
| SGLMM | GEA based on allele-environment association analysis | Landscape genomics | [222] | - |
| BayPass | GEA corrected for the covariance structure among the population allele frequencies | Landscape genomics | [224] | http://www1.montpellier.inra.fr/CBGP/software/baypass/ |
| BAYESCENV | GEA based on FST genome-scan | Landscape genomics | [225] | https://github.com/devillemereuil/bayescenv |
| LOSITAN | FST | Landscape genomics | [227] | https://mybiosoftware.com/lositan-1-0-0-selection-detection-workbench.html |
| PCAdmix | Supervised LAI | Local Ancestry Inference | [186] | https://sites.google.com/site/pcadmix/home |
| Tractor | LA-aware regression model | Local Ancestry Inference | [187] | https://github.com/eatkinson/Tractor |
| LAMP | LAI accounting for recombination | Local Ancestry Inference | [188] | http://lamp.icsi.berkeley.edu/lamp/ |
| MOSAIC (R package) | Unsupervised LAI | Local Ancestry Inference | [193] | https://maths.ucd.ie/~mst/MOSAIC/ |
| RFMix | LAI based on conditional random field | Local Ancestry Inference | [194] | https://github.com/slowkoni/rfmix |
| Loter | LAI for species other than humans | Local Ancestry Inference | [195] | https://github.com/bcm-uga/Loter |
| GHap (R package) | Unsupervised LAI | Local Ancestry Inference | [196] | https://cran.r-project.org/package=GHap |
| PSIKO2 | Unsupervised LAI | Local Ancestry Inference | [197] | https://www.uea.ac.uk/computing/psiko |
| SWIF(r) | Probabilistic method to detect selective sweeps | Deep Learning | [237] | https://github.com/ramachandran-lab/SWIFr |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Masson-Delmotte, V.; Zhai, P.; Pörtner, H.O.; Roberts, D.; Skea, J.; Shukla, P.R.; Pirani, A.; Moufouma-Okia, W.; Péan, C.; Pidcock, R.; et al. IPCC, 2018: Global Warming of 1.5 °C. An IPCC Special Report on the Impacts of Global Warming of 1.5 °C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate change, sustainable development, and efforts to eradicate poverty. 2018, in press. [Google Scholar]
- Ames, D.R.; Insley, L.W. Wind-Chill Effect for Cattle and Sheep. J. Anim. Sci. 1975, 40, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Toghiani, S.; Hay, E.H.; Roberts, A.; Rekaya, R. Impact of Cold Stress on Birth and Weaning Weight in a Composite Beef Cattle Breed. Livest. Sci. 2020, 236, 104053. [Google Scholar] [CrossRef]
- Pezzopane, R.M.; Carlos, A.; Bernardi, C.; Azenha, M.V.; Anch, P.P.; Bosi, C.; Pedroso, F.; Esteves, N. Production and Nutritive Value of Pastures in Integrated Livestock Production Systems: Shading and Management Effects. Sci. Agric. 2020, 77, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Belasco, E.; Cheng, Y.; Schroeder, T.C. The Impact of Extreme Weather on Cattle Feeding Profits. J. Agric. Resour. Econ. 2015, 40, 285–305. [Google Scholar]
- Lees, A.M.; Sejian, V.; Wallage, A.L.; Steel, C.C.; Mader, T.L.; Lees, J.C.; Gaughan, J.B. The Impact of Heat Load on Cattle. Animals 2019, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Rust, W.; Holman, I.; Bloomfield, J.; Cuthbert, M.; Corstanje, R. Understanding the Potential of Climate Teleconnections to Project Future Groundwater Drought. Hydrol. Earth Syst. Sci. 2019, 23, 3233–3245. [Google Scholar] [CrossRef] [Green Version]
- Thornton, P.K.; van de Steeg, J.; Notenbaert, A.; Herrero, M. The Impacts of Climate Change on Livestock and Livestock Systems in Developing Countries: A Review of What We Know and What We Need to Know. Agric. Syst. 2009, 101, 113–127. [Google Scholar] [CrossRef]
- Rotter, R.; Van de Geijn, S.C. Climate Change Effects On Plant Growth, Crop Yield and Livestock. Clim. Chang. 1999, 43, 651–681. [Google Scholar] [CrossRef]
- Wheeler, T.; Reynolds, C. Predicting the Risks from Climate Change to Forage and Crop Production for Animal Feed. Anim. Front. 2013, 3, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Bobea, A.; Knippenberg, E.; Chambers, R.G. Growing Climatic Sensitivity of U.S. Agriculture Linked to Technological Change and Regional Specialization. Sci. Adv. 2018, 4, eaat4343. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, J.B.; Cawdell-Smith, A.J. Impact of climate change on livestock production and reproduction. In Climate Change Impacts on Livestock: Adaptation and Mitigation; Sejian, V., Gaughan, J., Baumgard, L., Prasad, C., Eds.; Springer India: New Delhi, India, 2017; pp. 51–60. [Google Scholar]
- Hahn, G.; Mader, T.; Spiers, D.; Gaughan, J.; Nienaber, J.; Eigenberg, R.; Brown-Brandl, T.; Hu, Q.; Griffin, D.; Hugenford, L. Heat Wave Impacts on Feedlot Cattle: Considerations for Improved Environmental Management. In Proceedings of the Livestock Environment International Symposium Proceedings, Louisville, KY, USA, 21–23 May 2001. [Google Scholar]
- Mader, T.; Davis, S.; Gaughan, J.; Brown-brandl, T. Wind Speed and Solar Radiation Adjustments for the Temperature-Humidity Index. In Proceedings of the 16th Conference on Biometeorology and Aerobiology, Vancouver, BC, Canada, 23–27 August 2004; pp. 1–6. [Google Scholar]
- National Research Council Effect of enviroment on utrient requirement of domestic animals. In Subcommittee on Environmental Stress; National Research Council (Ed.) The National Academies Press: Washington, DC, USA, 1981. [Google Scholar]
- Hahn, G. Global Warming and Potential Impacts on Cattle and Swine in Tropical and Temperate Areas. In Proceedings of the Brazilian Congress of Biometeorology, USDA, Ljubljana, Slovenia, 1–8 September 1996. [Google Scholar]
- Thom, E.C. The Discomfort Index. Weatherwise 1959, 12, 57–61. [Google Scholar] [CrossRef]
- Amundson, J.L.; Mader, T.L.; Rasby, R.J.; Hu, Q.S. Environmental Effects on Pregnancy Rate in Beef Cattle 1. J. Anim. Sci. 2006, 84, 3415–3420. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.R.; Skea, J.; Buendia, E.C.; Masson-Delmotte, V.; Pörtner, H.-O.; Roberts, D.C.; Zhai, P.; Slade, R.; Connors, S.; van Diemen, R.; et al. IPCC, 2019: Climate Change and Land: An IPCC Special Report on Climate Change, Desertification, Land Degradation, Sustainable Land Management, Food Security, and Greenhouse Gas Fluxes in Terrestrial Ecosystems. 2019, in press. [Google Scholar]
- Lean, J.L.; Rind, D.H. How Will Earth’s Surface Temperature Change in Future Decades? Geophys. Res. Lett. 2009, 36, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Chapman, S.C.; Chakraborty, S.; Dreccer, F.; Howden, M. Plant Adaptation to Climate Change—Opportunities and Priorities in Breeding. Crop Pasture Sci. 2012, 63, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Calleja-Cabrera, J.; Boter, M.; Oñate-Sánchez, L.; Pernas, M. Root Growth Adaptation to Climate Change in Crops. Front. Plant Sci. 2020, 11, 544. [Google Scholar] [CrossRef]
- Tubiello, F.N.; Soussana, J.F.; Howden, S.M. Crop and Pasture Response to Climate Change. Proc. Natl. Acad. Sci. USA 2007, 104, 19686–19690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidosa, D.; Guyo, M. Journal of Fisheries & Climate Change Effects on Livestock Feed Resources: A Review. J. Fish. Livest. Prod. 2017, 5, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Morton, J.F. The Impact of Climate Change on Smallholder and Subsistence Agriculture. Proc. Natl. Acad. Sci. USA 2007, 104, 19680–19685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Mara, F.P. The Role of Grasslands in Food Security and Climate Change. Ann. Bot. 2012, 110, 1263–1270. [Google Scholar] [CrossRef] [Green Version]
- Baylis, M.; Risley, C. Infectious Diseases, Climate Change Effects on. In Infectious Diseases; Kanki, P., Grimes, D., Eds.; Springer: New York, NY, USA, 2013; pp. 117–146. [Google Scholar]
- Baylis, M.; Githeko, A.K. The Effects of Climate Change on Infectious Diseases of Animals; Report for the Foresight Project on Detection of Infectious Diseases; Department of Trade and Industry, UK Government: London, UK, 2006.
- Wilson, A.J.; Mellor, P.S. Bluetongue in Europe: Past, Present and Future. Philos. Trans. R. Soc. London. B 2009, 364, 2669–2681. [Google Scholar] [CrossRef]
- Olwoch, J.M.; Reyers, B.; Engelbrecht, F.A.; Erasmus, B.F.N. Climate Change and the Tick-Borne Disease, Theileriosis (East Coast Fever) in Sub-Saharan Africa. J. Arid Environ. 2008, 72, 108–120. [Google Scholar] [CrossRef]
- Kenyon, F.; Sargison, N.D.; Skuce, P.J.; Jackson, F. Sheep Helminth Parasitic Disease in South Eastern Scotland Arising as a Possible Consequence of Climate Change. Vet. Parasitol. 2009, 163, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.J.; Glenn, M.; Davidson, R.S.; White, P.C.L.; Hutchings, M. Climate-Driven Tipping-Points Could Lead to Sudden, High-Intensity Parasite Outbreaks. R. Soc. Open Sci. 2015, 2, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, C.L.; Smythe, L.D.; Craig, S.B.; Weinstein, P. Climate Change, Flooding, Urbanisation and Leptospirosis: Fuelling the Fire? Trans. R. Soc. Trop. Med. Hyg. 2010, 104, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Downing, M.M.; Nejadhashemi, A.P.; Harrigan, T.; Woznicki, S.A. Climate Change and Livestock: Impacts, Adaptation, and Mitigation. Clim. Risk Manag. 2017, 16, 145–163. [Google Scholar] [CrossRef]
- Buontempo, C.; Hutjes, R.; Beavis, P.; Berckmans, J.; Cagnazzo, C.; Vamborg, F.; Bergeron, C.; Almond, S.; Amici, A.; Ramasamy, S.; et al. Fostering the Development of Climate Services through Copernicus Climate Change Service (C3S) for Agriculture Applications. Weather Clim. Extrem. 2020, 27, 1–8. [Google Scholar] [CrossRef]
- Fick, S.E.; Hijmans, R.J. WorldClim 2: New 1-Km Spatial Resolution Climate Surfaces for Global Land Areas. Int. J. Climatol. 2017, 37, 4302–4315. [Google Scholar] [CrossRef]
- Karger, D.N.; Conrad, O.; Böhner, J.; Kawohl, T.; Kreft, H.; Soria-auza, R.W.; Zimmermann, N.E.; Linder, H.P.; Kessler, M. Data Descriptor: Climatologies at High Resolution for the Earth’s Land Surface Areas. Sci. Data 2017, 4, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kriticos, D.J.; Webber, B.L.; Leriche, A.; Ota, N.; Macadam, I.; Bathols, J.; Scott, J.K. CliMond: Global High-Resolution Historical and Future Scenario Climate Surfaces for Bioclimatic Modelling. Methods Ecol. Evol. 2012, 3, 53–64. [Google Scholar] [CrossRef]
- Lima-Ribeiro, M.S.; Varela, S.; González-Hernández, J.; de Oliveira, G.; Diniz-Filho JA, F.; Terribile, L.C. EcoClimate: A Database of Climate Data from Multiple Models for Past, Present, and Future for Macroecologists and Biogeographers. Biodivers. Inform. 2015, 10, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Title, P.O.; Bemmels, B. ENVIREM: An Expanded Set of Bioclimatic and Topographic Variables Increases Flexibility and Improves Performance of Ecological Niche Modeling. Ecography 2018, 41, 291–307. [Google Scholar] [CrossRef] [Green Version]
- Vega, G.C.; Pertierra, L.R.; Olalla-tárraga, M.Á. Data Descriptor: MERRAclim, a High-Resolution Global Dataset of Remotely Sensed Bioclimatic Variables for Ecological Modelling. Sci. Data 2017, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noce, S.; Caporaso, L.; Santini, M. A New Global Dataset of Bioclimatic Indicators. Sci. Data 2020, 7, 1–12. [Google Scholar] [CrossRef]
- Cui, D.; Liang, S.; Wang, D.; Liu, Z. Köppen-Geiger Climate Classification and Bioclimatic Variables. Earth Syst. Sci. Data 2021, 1–34. [Google Scholar] [CrossRef]
- Thépaut, J.; Dee, D.; Engelen, R.; Pinty, B. The Copernicus Programme and Its Climate Change Service. In Proceedings of the IGARSS 2018—2018 IEEE International Geoscience and Remote Sensing Symposium, Valencia, Spain, 22–27 July 2018; pp. 1591–1593. [Google Scholar]
- Sillmann, J.; Kharin, V.V.; Zhang, X.; Zwiers, F.W.; Bronaugh, D. Climate Extremes Indices in the CMIP5 Multimodel Ensemble: Part 1. Model Evaluation in the Present Climate. J. Geophys. Res. 2013, 118, 1716–1733. [Google Scholar] [CrossRef]
- Sillmann, J.; Kharin, V.V.; Zwiers, F.W.; Zhang, X.; Bronaugh, D. Climate Extremes Indices in the CMIP5 Multimodel Ensemble: Part 2. Future Climate Projections. J. Geophys. Res. 2013, 118, 2473–2493. [Google Scholar] [CrossRef]
- Naderi, S.; Rezaei, H.R.; Taberlet, P.; Zundel, S.; Rafat, S.; Naghash, H.; El-Barody, M.A.A.; Ertugrul, O.; Pompanon, F. Large-Scale Mitochondrial DNA Analysis of the Domestic Goat Reveals Six Haplogroups with High Diversity. PLoS ONE 2007, 2, e1012. [Google Scholar] [CrossRef] [Green Version]
- Zeder, M.A. Domestication and Early Agriculture in the Mediterranean Basin: Origins, Diffusion, and Impact. Proc. Natl. Acad. Sci. USA 2008, 19, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Zeder, M.A. Core Questions in Domestication Research. Proc. Natl. Acad. Sci. USA 2015, 17, 3192–3198. [Google Scholar] [CrossRef] [Green Version]
- Legge, T. The beginnings of caprine domestication. In The Origins and Spread of Agriculture and Pastoralism in Eurasia; Harris, D.R., Ed.; Smithsonian Institution Press: New York, NY, USA, 1996; pp. 238–262. [Google Scholar]
- Vigne, J. The Origins of Animal Domestication and Husbandry: A Major Change in the History of Humanity and the Biosphere. Comptes Rendus Biol. 2011, 334, 171–181. [Google Scholar] [CrossRef]
- Mchugo, G.P.; Dover, M.J.; Machugh, D.E. Unlocking the Origins and Biology of Domestic Animals Using Ancient DNA and Paleogenomics. BMC Biol. 2019, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.D.E.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome Sequencing of the Extinct Eurasian Wild Aurochs, Bos Primigenius, Illuminates the Phylogeography and Evolution of Cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, K.G.; Delser, P.M.; Mullin, V.E.; Scheu, A.; Mattiangeli, V.; Teasdale, M.D.; Hare, A.J.; Burger, J.; Verdugo, M.P.; Collins, M.J.; et al. Ancient Goat Genomes Reveal Mosaic Domestication in the Fertile Crescent. Science 2018, 361, 24–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdugo, M.P.; Mullin, V.E.; Scheu, A.; Mattiangeli, V.; Daly, K.G.; Delser, P.M.; Hare, A.J.; Burger, J.; Collins, M.J.; Kehati, R.; et al. Ancient Cattle Genomics, Origins and Rapid Turnover in the Fertile Crescent. Science 2019, 176, 173–176. [Google Scholar]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Guo, Y.; et al. The Origin of Domestication Genes in Goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Gamba, C.; Gaunitz, C.; Der Sarkissian, C.; Pruvost, M.; Albrechtsen, A.; Fages, A.; Khan, N.; Schubert, M.; Jagannathan, V.; et al. Ancient Genomic Changes Associated with Domestication of the Horse. Science 2017, 356, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, A.; Shen, F.; Taravella, A.; Emery, S.; Veeramah, K.; Boyko, A.; Kidd, J.M. Comparison of Village Dog and Wolf Ge-Nomes Highlights the Role of the Neural Crest in Dog Domestication. BMC Biol. 2018, 16, 64. [Google Scholar] [CrossRef] [Green Version]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 Systems—Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta—Gen. Subj. 2007, 1770, 330–344. [Google Scholar] [CrossRef]
- Weinberg, P.J. Capra Cylindricornis. Mamm. Species 2002, 695, 1–9. [Google Scholar] [CrossRef]
- Liu, Y.; Weyrich, L.; Llamas, B. More Arrows in the Ancient DNA Quiver: Use of Paleoepigenomes and Paleomicrobiomes to Investigate Animal Adaptation to Environment. Mol. Biol. Evol. 2020, 37, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Baumgard, L.H.; Zimbelman, R.B.; Xiao, Y. Heat Stress: Physiology of Acclimation and Adaptation. Anim. Front. 2019, 9, 12–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaughan, J.B.; Sejian, V.; Mader, T.L.; Dunshea, F.R. Adaptation Strategies: Ruminants. Anim. Front. 2019, 9, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignon-grasteau, S.; Boissy, A.; Bouix, J.; Faure, J.; Fisher, A.D.; Hinch, G.N.; Jensen, P.; Le, P.; Prunet, P.; Vandeputte, M.; et al. Genetics of Adaptation and Domestication in Livestock B. Livest. Prod. Sci. 2005, 93, 3–14. [Google Scholar] [CrossRef]
- Galal, S.; Rasoul, F.A.; Annous, M.R.; Shoat, I. Small Ruminant Breeds of Egypt; Aleppo, 2005; International Center for Agricultural Research in Dry Areas (ICARDA): Aleppo, Syria, 2005; 196p. [Google Scholar]
- Mazzullo, G.; Rifici, C.; Cammarata, F.; Caccamo, G.; Rizzo, M.; Piccione, G.; Practician, V.; Practicioner, V. Effect of Different Environmental Conditions on Some Haematological Parameters in Cow. Ann. Anim. Sci. 2014, 14, 947–954. [Google Scholar] [CrossRef] [Green Version]
- Hansen, P. Physiological and Cellular Adaptations of Zebu Cattle to Thermal Stress. Anim. Reprod. Sci. 2004, 82–83, 349–360. [Google Scholar] [CrossRef]
- Utsunomiya, Y.T.; Milanesi, M.; Fortes, M.R.S. Genomic Clues of the Evolutionary History of Bos Indicus Cattle. Anim. Genet. 2019, 50, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Trail, J.C.M.; Gregory, K.E.; Marples, H.J.S.; Kakonge, J. Comparison of Bos Taurus-Bos Indicus Breed Crosses with Straightbred Bos Indicus Breeds of Cattle for Maternal and Individual Traits. J. Anim. Sci. 1985, 60, 1181–1187. [Google Scholar] [CrossRef]
- Madalena, F.E. DAIRY ANIMALS|Bos Indicus Breeds and Bos Indicus × Bos Taurus Crosses. Encycl. Dairy Sci. 2002, 576–585. [Google Scholar] [CrossRef]
- Hill, D.; Wall, E. Dairy Cattle in a Temperate Climate: The Effects of Weather on Milk Yield and Composition Depend on Management. Animal 2015, 9, 138–149. [Google Scholar] [CrossRef] [Green Version]
- da Silva, V.P.R.; e Silva, R.A.; Cavalcanti, E.P.; Braga, C.C.; de Azevedo, P.V.; Singh, V.P.; Pereira, E.R.R. Trends in Solar Radiation in NCEP/NCAR Database and Measurements in Northeastern Brazil. Sol. Energy 2010, 84, 1852–1862. [Google Scholar] [CrossRef]
- Herbut, P.; Angrecka, S.; Walczak, J. Environmental Parameters to Assessing of Heat Stress in Dairy Cattle—A Review. Int. J. Biometeorol. 2018, 62, 2089–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, N.R.; Cobanov, B.; Schnitkey, G. Economic Losses from Heat Stress by US Livestock Industries. J. Dairy Sci. 2003, 86, E52–E77. [Google Scholar] [CrossRef] [Green Version]
- Baêta, F.C.; Meador, N.; Shanklin, M.D.; Johnson, H.D. Equivalent Temperature Index at Temperatures above the Thermoneu-Tral for Lactating Dairy Cows. In Proceedings of the Meeting of the American Society of Agricultural engineers, Chicago, IL, USA, 1987; Available online: https://agris.fao.org/agris-search/search.do?recordID=US8853966 (accessed on 21 July 2021).
- Mader, T.L.; Davis, M.S.; Brown-Brandl, T. Environmental Factors Influencing Heat Stress in Feedlot Cattle. J. Anim. Sci. 2006, 84, 712–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eigenberg, R.A.; Brown-Brandl, T.M.; Nienaber, J.A.; Hahn, G.L. Dynamic Response Indicators of Heat Stress in Shaded and Non-Shaded Feedlot Cattle, Part 2: Predictive Relationships. Biosyst. Eng. 2005, 91, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Rashamol, V.P.; Sejian, V.; Pragna, P.; Lees, A.M.; Bagath, M.; Krishnan, G.; Gaughan, J.B. Prediction Models, Assessment Methodologies and Biotechnological Tools to Quantify Heat Stress Response in Ruminant Livestock. Int. J. Biometeorol. 2019, 63, 1265–1281. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, J.B.; Mader, T.L.; Holt, S.M.; Lisle, A. A New Heat Load Index for Feedlot Cattle. J. Anim. Sci. 2008, 86, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Mader, T.L.; Johnson, L.J.; Gaughan, J.B.; Mader, T.L.; Johnson, L.J.; Gaughan, J.B. A Comprehensive Index for Assessing Environmental Stress in Animals. J. Anim. Sci. 2010, 88, 2153–2165. [Google Scholar] [CrossRef] [Green Version]
- Flori, L.; Gonzatti, M.I.; Thevenon, S.; Chantal, I.; Pinto, J.; Berthier, D.; Aso, P.M.; Gautier, M. A Quasi-Exclusive European Ancestry in the Senepol Tropical Cattle Breed Highlights the Importance of the Slick Locus in Tropical Adaptation. PLoS ONE 2012, 7, e36133. [Google Scholar] [CrossRef]
- Olson, T.A.; Lucena, C.; Chase, C.C.J.; Hammond, A.C. Evidence of a Major Gene Influencing Hair Length and Heat Tolerance in Bos Taurus Cattle. J. Anim. Sci. 2003, 81, 80–90. [Google Scholar] [CrossRef]
- Dikmen, S.; Alava, E.; Pontes, E.; Fear, J.M.; Dikmen, B.Y.; Olson, T.A.; Hansen, P.J. Differences in Thermoregulatory Ability Between Slick-Haired and Wild-Type Lactating Holstein Cows in Response to Acute Heat Stress. J. Dairy Sci. 2008, 91, 3395–3402. [Google Scholar] [CrossRef] [PubMed]
- Mariasegaram, M.; Chase, C.C.J.; Chaparro, J.X.; Olson, T.A.; Brenneman, R.A.; Niedz, R.P. The Slick Hair Coat Locus Maps to Chromosome 20 in Senepol-Derived Cattle. Anim. Genet. 2007, 38, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Littlejohn, M.D.; Henty, K.M.; Tiplady, K.; Johnson, T.; Harland, C.; Lopdell, T.; Sherlock, R.G.; Li, W.; Lukefahr, S.D.; Shanks, B.C.; et al. Functionally Reciprocal Mutations of the Prolactin Signalling Pathway Define Hairy and Slick Cattle. Nat. Commun. 2014, 5, 5861. [Google Scholar] [CrossRef] [Green Version]
- Martinez, A.M.; Gama, L.T.; Canon, J.; Ginja, C.; Delgado, J.V.; Dunner, S.; Al, E. Genetic Footprints of Iberian Cattle in America 500 Years after the Arrival of Columbus. PLoS ONE 2012, 7, e49066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto-neto, L.R.; Bickhart, D.M.; Landaeta-hernandez, A.J.; Utsunomiya, Y.T.; Pagan, M.; Jimenez, E.; Hansen, P.J.; Dikmen, S.; Schroeder, S.G.; Kim, E.; et al. Convergent Evolution of Slick Coat in Cattle through Truncation Mutations in the Prolactin Receptor. Front. Genet. 2018, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Mundel, T.; Bunn, S.J.; Hooper, P.L.; Jones, D.A. The Effects of Face Cooling during Hyperthermic Exercise in Man: Evidence for an Integrated Thermal, Neuroendocrine and Behavioural Response. Exp. Physiol. 2007, 92, 187–195. [Google Scholar] [CrossRef]
- Dikmen, S.; Khan, F.A.; Huson, H.J.; Sonstegard, T.S.; Moss, J.I.; Dahl, G.E.; Hansen, P.J. The SLICK Hair Locus Derived from Senepol Cattle Confers Thermotolerance to Intensively Managed Lactating Holstein Cows. J. Dairy Sci. 2014, 97, 5508–5520. [Google Scholar] [CrossRef] [Green Version]
- Bahbahani, H.; Tijjani, A.; Mukasa, C.; Wragg, D. Signatures of Selection for Environmental Adaptation and Zebu × Taurine Hybrid Fitness in East African Shorthorn Zebu. Front. Genet. 2017, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. International Human Genome Consortium Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and Comparative Analysis of the Chicken Genome Provide Unique Perspectives on Vertebrate Evolution. Nature 2004, 432, 695–777. [Google Scholar] [CrossRef]
- The Bovine genome Sequencing and Analysis Consortium. The Genome Sequence of Taurine Cattle: A Window to Ruminant Biology and Evolution. Science 2009, 324, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Groenen, M.A.M.; Archibald, A.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gailard, C.; Park, C.; Milan, D.; Megens, H.; et al. Analyses of Pig Genomes Provide Insight into Porcine Demography and Evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The Sheep Genome Illuminates Biology of the Rumen and Lipid Metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.; Du, X.; Zhang, W.; Tosser-klopp, G.; Wang, J.; Yang, S.; Liang, J.; et al. Sequencing and Automated Whole-Genome Optical Mapping of the Genome of a Domestic Goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Metzker, M.L. Sequencing Technologies—The next Generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.P.; Kearney, J.F. Imputation of Genotypes from Low- to High-Density Genotyping Platforms and Implications for Genomic Selection. Anim. Int. J. Anim. Biosci. 2011, 5, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Vanvanhossou, S.F.U.; Scheper, C.; Dossa, L.H.; Yin, T.; Brügemann, K.; König, S. A Multi-Breed GWAS for Morphometric Traits in Four Beninese Indigenous Cattle Breeds Reveals Loci Associated with Conformation, Carcass and Adaptive Traits. BMC Genom. 2020, 21, 783. [Google Scholar] [CrossRef] [PubMed]
- Igoshin, A.V.; Yurchenko, A.A.; Belonogova, N.M.; Petrovsky, D.V.; Aitnazarov, R.B.; Soloshenko, V.A.; Yudin, N.S.; Larkin, D.M. Genome-Wide Association Study and Scan for Signatures of Selection Point to Candidate Genes for Body Temperature Maintenance under the Cold Stress in Siberian Cattle Populations. BMC Genet. 2019, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Porto-neto, L.R.; Reverter, A.; Prayaga, K.C. The Genetic Architecture of Climatic Adaptation of Tropical Cattle. PLoS ONE 2014, 9, e113284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raven, L.-A.; Cocks, B.G.; Hayes, B.J. Multibreed Genome Wide Association Can Improve Precision of Mapping Causative Variants Underlying Milk Production in Dairy Cattle. BMC Genomics 2014, 15, 62. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Kijas, J.W.; Townley, D.; Dalrymple, B.P.; Heaton, M.P.; Maddox, J.F.; Wilson, P.; Ingersoll, R.G.; Mcculloch, R.; Mcwilliam, S.; Tang, D.; et al. A Genome Wide Survey of SNP Variation Reveals the Genetic Structure of Sheep Breeds. PLoS ONE 2009, 4, e4668. [Google Scholar] [CrossRef] [Green Version]
- Michailidou, S.; Tsangaris, G.T.; Tzora, A.; Skoufos, I.; Banos, G.; Argiriou, A.; Arsenos, G. Analysis of Genome-Wide DNA Arrays Reveals the Genomic Population Structure and Diversity in Autochthonous Greek Goat Breeds. PLoS ONE 2019, 14, e0226179. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Cui, L.; Enciso, M.P.; Traspov, A.; Crooijmans, R.P.M.A.; Zinovieva, N.; Schook, L.B.; Archibald, A.; Gatphayak, K.; Knorr, C. Genome-Wide SNP Data Unveils the Globalization of Domesticated Pigs. Genet. Sel. Evol. 2017, 49, 71. [Google Scholar] [CrossRef] [Green Version]
- Bruford, M.W.; Ginja, C.; Hoffmann, I.; Joost, S.; Orozco-terWengel, P.; Alberto, F.J.; Amaral, A.J.; Barbato, M.; Biscarini, F.; Colli, L.; et al. Prospects and Challenges for the Conservation of Farm Animal Genomic Resources, 2015–2025. Front. Genet. 2015, 6, 314. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Bickhart, D.M.; Cole, J.B.; Schroeder, S.G.; Song, J.; Van Tassell, C.P.; Sonstegard, T.; Liu, G.E. Genomic Signatures Reveal New Evidences for Selection of Important Traits in Domestic Cattle. Mol. Biol. Evol. 2015, 32, 711–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, I.; Kim, S. Analysis of Whole Transcriptome Sequencing Data: Workflow and Software. Genom. Inform. 2015, 13, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Capoferri, R.; Panelli, S.; Minozzi, G.; Strozzi, F.; Trevisi, E.; Snel, G.G.M.; Ajmone-Marsan, P.; Williams, J.L. Johne’s Disease in Cattle: An in Vitro Model to Study Early Response to Infection of Mycobacterium Avium Subsp Paratuberculosis Using RNA-Seq. Mol. Immunol. 2016, 91, 259–271. [Google Scholar] [CrossRef]
- Wang, L.; Cai, B.; Zhou, S.; Zhu, H.; Qu, L.; Wang, X.; Al, E. RNA-Seq Reveals Transcriptome Changes in Goats Following Myostatin Gene Knockout. PLoS ONE 2017, 12, e0187966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zappaterra, M.; Gioiosa, S.; Chillemi, G.; Zambonelli, P.; Davoli, R. Muscle Transcriptome Analysis Identifies Genes Involved in Ciliogenesis and the Molecular Cascade Associated with Intramuscular Fat Content in Large White Heavy Pigs. PLoS ONE 2020, 15, e0233372. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Chitwood, J. Genome-Wide Identification of Tissue-Specific Long Non-Coding RNA in Three Farm Animal Species. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, X.; Luo, Q.; Zhao, H.; Al, E. Co-Expression Analysis and Identification of Fecundity-Related Long Non-Coding RNAs in Sheep Ovaries. Sci. Rep. 2016, 6, 39398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquariello, R.; Fernandez-Fuertes, B.; Strozzi, F.; Mazza, R.; Lonergan, P.; Gandolfi, F.; Williams, J.L. Profiling Bovine Blastocyst MicroRNAs Using Deep Sequencing. J. Reprod. Fertil. Dev. 2017, 29, 1545–1555. [Google Scholar] [CrossRef]
- Low, W.Y.; Tearle, R.; Liu, R.; Koren, S.; Rhie, A.; Bickhart, D.M.; Rosen, B.D.; Kronenberg, Z.N.; Kingan, S.B.; Tseng, E.; et al. Haplotype-Resolved Genomes Provide Insights into Structural Variation and Gene Content in Angus and Brahman Cattle. Nat. Commun. 2020, 11, 2071. [Google Scholar] [CrossRef]
- Koren, S.; Rhie, A.; Walenz, B.P.; Dilthey, A.T.; Bickhart, D.M.; Kingan, S.B.; Hiendleder, S.; Williams, J.L.; Smith, T.P.L.; Adam, M. Complete Assembly of Parental Haplotypes with Trio Binning. bioRxiv 2018, 271486. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Mao, K.; Li, J.; Huang, W.; Che, T.; Fu, Y.; Tang, Q.; Liu, P.; Song, Y.; Liu, R.; et al. Genome-Wide Profiling of Gene Expression and DNA Methylation Provides Insight into Low-Altitude Acclimation in Tibetan Pigs. Gene 2018, 5, 522–532. [Google Scholar] [CrossRef]
- Giuffra, E.; Tuggle, C.K.; FAANG Consortium. Functional Annotation of Animal Genomes (FAANG): Current Achievements and Roadmap. Annu. Rev. Anim. Biosci. 2018, 7, 65–88. [Google Scholar] [CrossRef]
- Georges, M.; Charlier, C.; Hayes, B. Harnessing Genomic Information for Livestock Improvement. Nat. Rev. Genet. 2019, 20, 135–156. [Google Scholar] [CrossRef]
- Cantor, R.M.; Lange, K.; Sinsheimer, J.S. Prioritizing GWAS Results: A Review of Statistical Methods and Recommendations for Their Application. Am. J. Hum. Genet. 2010, 86, 6–22. [Google Scholar] [CrossRef] [Green Version]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef]
- Hatzikotoulas, K.; Gilly, A.; Zeggini, E. Using Population Isolates in Genetic Association Studies. Brief. Funct. Genom. 2014, 13, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Seop, J.; Dang, C.G.; Sudrajad, P.; Kim, H.C. Stories and Challenges of Genome Wide Association Studies in Livestock—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Lewin, H.A.; Goddard, M.E. The Future of Livestock Breeding: Genomic Selection for Efficiency, Reduced Emissions Intensity, and Adaptation. Trends Genet. 2013, 29, 206–214. [Google Scholar] [CrossRef]
- Hayes, B. Overview of Statistical Methods for Genome-Wide Association Studies (GWAS). Methods Mol. Biol. 2013, 1019, 149–169. [Google Scholar] [PubMed]
- Goddard, M.E.; Hayes, B.J. Mapping Genes for Complex Traits in Domestic Animals and Their Use in Breeding Programmes. Nat. Rev. 2009, 10, 381–391. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-Analysis of Genome-Wide Association Studies for Cattle Stature Identifies Common Genes That Regulate Body Size in Mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef]
- Kominakis, A.; Hager-Theodorides, A.L.; Zoidis, E.; Saridaki, A.; Antonakos, G.; Tsiamis, G. Combined GWAS and ‘Guilt by Association’-Based Prioritization Analysis Identifies Functional Candidate Genes for Body Size in Sheep. Genet. Sel. Evol. 2017, 49, 41. [Google Scholar] [CrossRef]
- Braz, C.U.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E. Genome-wide Association Analyses Identify Genotype-by-environment Interactions of Growth Traits in Simmental Cattle. Sci. Rep. 2021, 11, 13335. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.M.; Swiger, L.; Chambers, D.; Gregory, K. Efficiency of Feed Use in Beef Cattle. J. Anim. Sci. 1963, 22, 486–494. [Google Scholar] [CrossRef]
- Herd, R.M.; Bishop, S.C. Genetic Variation in Residual Feed Intake and Its Association with Other Production Traits in British Hereford Cattle. Livest. Prod. Sci. 2000, 63, 111–119. [Google Scholar] [CrossRef]
- Tortereau, F.; Weisbecker, J.; Marcon, D.; Bouvier, F.; François, D. Genetic Parameters for Feed Efficiency in Romane Rams and Responses to Single-Generation Selection. Anim. Int. J. Anim. Biosci. 2020, 14, 681–687. [Google Scholar] [CrossRef]
- Santana, M.H.A.; Utsunomiya, Y.T.; Neves, H.H.R.; Gomes, R.C.; Garcia, J.F.; Fukumasu, H.; Silva, S.L.; Oliveira Junior, G.A.; Alexandre, P.A.; Leme, P.R.; et al. Genome-Wide Association Analysis of Feed Intake and Residual Feed Intake in Nellore Cattle. BMC Genet. 2014, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, P.S.N.; Cesar, A.S.M.; Nascimento, M.L.; Chaves, A.S.; Tizioto, P.C.; Tullio, R.R.; Lanna, D.P.D.; Rosa, A.N.; Sonstegard, T.S.; Mourao, G.B.; et al. Identification of Genomic Regions Associated with Feed Efficiency in Nelore Cattle. BMC Genet. 2014, 15, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, D.G.; Gill, C.A.; Boldt, C.R.; Funkhouser, R.R.; Herring, A.D.; Riggs, P.K.; Sawyer, J.E.; Lunt, D.K.; Sanders, J.O. Crossbred Steer Temperament as Yearlings and Whole Genome Association of Steer Temperament as Yearlings and Calf Temperament Post-Weaning. J. Anim. Sci. 2016, 94, 1408–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, A.; Daures, J.; van Arendonk, J.A.M.; Bovenhuis, H. Genome-Wide Association Study for Behavior, Type Traits, and Muscular Development in Charolais Beef Cattle. J. Anim. Sci. 2016, 94, 2307–2316. [Google Scholar] [CrossRef]
- Paredes-Sanchez, F.A.; Sifuentes-Rincon, A.M.; Casas, E.; Arellano-vera, W.; Parra-bracamonte, G.M.; Riley, D.G.; Welsh, T.H., Jr.; Randel, R.D. Novel Genes Involved in the Genetic Architecture of Temperament in Brahman Cattle. PLoS ONE 2020, 15, e0237825. [Google Scholar] [CrossRef]
- Vitti, J.J.; Grossman, S.R.; Sabeti, P.C. Detecting Natural Selection in Genomic Data. Annu. Rev. Genet. 2013, 47, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; De Vlaming, R.; Wu, Y.; Robinson, M.R.; Lloyd-Jones, L.R.; Yengo, L.; Yap, C.X.; Xue, A.; Sidorenko, J.; McRae, A.F.; et al. Signatures of Negative Selection in the Genetic Architecture of Human Complex Traits. Nat. Genet. 2018, 50, 746–753. [Google Scholar] [CrossRef]
- Charlesworth, D. Balancing Selection and Its Effects on Sequences in Nearby Genome Regions. PLoS Genet. 2006, 2, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Barton, N.H. Genetic Hitchhiking. Trans. R. Soc. B 2000, 355, 553–1562. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.H.; Xu, S.S.; Shen, M.; Chen, Z.H.; Gao, L.; Lv, F.H.; Xie, X.L.; Wang, X.H.; Yang, H.; Liu, C.-B.; et al. Historical Introgression from Wild Relatives Enhanced Climatic Adaptation and Resistance to Pneumonia in Sheep. Mol. Biol. Evol. 2021, 38, 838–855. [Google Scholar] [CrossRef] [PubMed]
- Yurchenko, A.A.; Daetwyler, H.D.; Yudin, N.; Schnabel, R.D.; Vander Jagt, C.J.; Soloshenko, V.; Lhasaranov, B.; Popov, R.; Taylor, J.F.; Larkin, D.M. Scans for Signatures of Selection in Russian Cattle Breed Genomes Reveal New Candidate Genes for Environmental Adaptation and Acclimation. Sci. Rep. 2018, 8, 12984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yang, Y.; Li, C.; Li, R.; Xiao, H.; Chen, S. Genetic Diversity of TLR3 and TLR8 Genes among Five Chinese Native Cattle Breeds from Southwest China. Livest. Sci. 2020, 232, 103895. [Google Scholar] [CrossRef]
- Eusebi, P.G.; Cortés, O.; Carleos, C.; Dunner, S.; Cañon, J. Detection of Selection Signatures for Agonistic Behaviour in Cattle. J. Anim. Breed. Genet. 2018, 135, 170–177. [Google Scholar] [CrossRef]
- Eusebi, P.G.; Sevane, N.; Cortés, O.; Contreras, E.; Cañon, J.; Dunner, S. Aggressive Behavior in Cattle Is Associated with a Polymorphism in the MAOA Gene Promoter. Anim. Genet. 2020, 51, 14–21. [Google Scholar] [CrossRef]
- Hamlyn-Hill, F. Improving Temperament: Effects on Productivity and Meat Quality. Beef CRC. Available online: http://futurebeef.com.au/topics/breeding-and-genetics/improving-temperament-andflight-time (accessed on 28 July 2021).
- Burdick, N.; Agado, B.; White, J.; Matheney, K.; Neuendorff, D.; Riley, D.; Vann, R.C.; Welsh, T.H., Jr.; Randel, R.D. Technical Note: Evolution of Exit Velocity in Suckling Brahman Calves. J. Anim. Sci. 2011, 89, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Café, L.M.; Robinson, D.L.; Ferguson, D.M.; McIntyre, B.L.; Geesink, G.H.; Greenwood, P.L. Cattle Temperament: Persistence of Assessments and Associations with Productivity, Efficiency, Carcass and Meat Quality Traits. J. Anim. Sci. 2011, 89, 1452–1465. [Google Scholar] [CrossRef]
- Burrow, H. Variances and Covariances between Productive and Adaptative Traits and Temperament in a Composite Breed of Tropical Beef Cattle. Livest. Prod. Sci. 2001, 70, 213–233. [Google Scholar] [CrossRef]
- Hoppe, S.; Brandt, H.R.; Nig, S.K.; Erhardt, G.; Gauly, M. Temperament Traits of Beef Calves Measured under Field Condi-Tions and Their Relationships to Performance. J. Anim. Sci. 2010, 88, 1892–1898. [Google Scholar] [CrossRef]
- Lv, F.; Agha, S.; Kantanen, J.; Colli, L.; Stucki, S.; Kijas, J.W.; Li, M.; Marsan, P.A. Adaptations to Climate-Mediated Selective Pressures in Sheep. Mol. Biol. Evol. 2014, 31, 3324–3343. [Google Scholar] [CrossRef] [Green Version]
- Igoshin, A.; Yudin, N.; Aitnazarov, R.; Yurchenko, A.A.; Larkin, D.M. Whole-Genome Resequencing Points to Candidate DNA Loci Affecting Body Temperature under Cold Stress in Siberian Cattle Populations. Life 2021, 11, 959. [Google Scholar] [CrossRef]
- Li, R.; Li, H.; Chen, R.; Chong, Q.; Xiao, H.; Chen, S. Genome-Wide Scan of Selection Signatures in Dehong Humped Cattle for Heat Tolerance and Disease Resistance. Anim. Genet. 2020, 51, 292–299. [Google Scholar]
- Edea, Z.; Dadi, H.; Dessie, T.; Kim, K.S. Genomic Signatures of High-Altitude Adaptation in Ethiopian Sheep Popula-Tions. Genes Genom. 2019, 41, 973–981. [Google Scholar] [CrossRef]
- Freitas, P.H.F.; Wang, Y.; Yan, P.; Oliveira, H.R.; Schenkel, F.S.; Zhang, Y.; Brito, L.F. Genetic Diversity and Signatures of Selection for Thermal Stress in Cattle and Other Two Bos Species Adapted to Divergent Climatic Conditions. Front. Genet. 2021, 12, 102. [Google Scholar] [CrossRef]
- Buggiotti, L.; Yurchenko, A.A.; Yudin, N.S.; Vander Jagt, C.J.; Nadezhda, V.; Kusliy, M.; Vasiliev, S.K.; Rodionov, A.N.; Boronetskaya, O.I.; Zinovieva, A.; et al. Demographic History, Adaptation, and NRAP Convergent Evolution at Amino Acid Residue 100 in the World Northernmost Cattle from Siberia. Mol. Biol. Evol. 2021, 38, 3093–3110. [Google Scholar] [CrossRef]
- Wang, X.; Ju, Z.; Jiang, Q.; Zhong, J.; Liu, C.; Wang, J.; Hoff, J.L.; Schnabel, R.D.; Zhao, H.; Gao, Y.; et al. Introgression, Admixture and Selection Facilitate Genetic Adaptation to High-Altitude Environments in Cattle. Genomics 2021, 113, 1491–1503. [Google Scholar] [CrossRef]
- Molotsi, A.; Cloete, S.; Taylor, J.F.; Whitacre, L. Identification of Selection Signatures in South African Sheep Popula-Tions Using HAPFLK and Bayesian Fst Approaches. In Proceedings of the World Congress on Genetics Applied to Livestock, Auckland, New Zealand, 7–11 February 2018; pp. 1–5. [Google Scholar]
- Mwacharo, J.M.; Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.A.; Rothschild, M.F. Genomic Footprints of Dryland Stress Adaptation in Egyptian Fat-Tail Sheep and Their Divergence from East African and Western Asia Cohorts. Sci. Rep. 2017, 7, 17647. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, K.A.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B.P. Genome-Wide Analysis of Genetic Diversity and Selection Signatures in Three Indian Sheep Breeds. Livest. Sci. 2021, 243, 104367. [Google Scholar] [CrossRef]
- Álvarez, I.; Fernández, I.; Traoré, A.; Pérez-Pardal, L.; Menéndez-Arias, N.A.; Goyache, F. Ancient Homozygosity Segments in West African Djallonké Sheep Inform on the Genomic Impact of Livestock Adaptation to the Environment. Animals 2020, 10, 1178. [Google Scholar] [CrossRef]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; Steri, R.; et al. Signatures of Selection and Environmental Adaptation across the Goat Genome Post-Domestication. Genet. Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef]
- Kim, E.S.; Elbeltagy, A.R.; Aboul-Naga, A.M.; Rischkowsky, B.; Sayre, B.; Mwacharo, J.M.; Rothschild, M.F. Multiple Genomic Signatures of Selection in Goats and Sheep Indigenous to a Hot Arid Environment. Heredity (Edinb) 2016, 116, 255–264. [Google Scholar] [CrossRef]
- Pitt, D.; Bruford, M.W.; Barbato, M.; Orozco-terWengel, P.; Martínez, R.; Sevane, N. Demography and Rapid Local Adaptation Shape Creole Cattle Genome Diversity in the Tropics. Evol. Appl. 2019, 12, 105–122. [Google Scholar] [CrossRef]
- Dutta, P.; Talenti, A.; Young, R.; Jayaraman, S.; Callaby, R.; Jadhav, S.K.; Dhanikachalam, V.; Manikandan, M.; Biswa, B.B.; Low, W.Y.; et al. Whole Genome Analysis of Water Buffalo and Global Cattle Breeds Highlights Convergent Signatures of Domestication. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Singh, A.; Mehrotra, A.; Gondro, C.; da Silva Romero, A.R.; Pandey, A.K.; Karthikeyan, A.; Bashir, A.; Mishra, B.P.; Dutt, T.; Kumar, A. Signatures of Selection in Composite Vrindavani Cattle of India. Front. Genet. 2020, 11, 589496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xue, X.; Liu, Y.; Abied, A.; Ding, Y.; Zhao, S.; Wang, W.; Ma, L.; Guo, J.; Guan, W.; et al. Genome-Wide Comparative Analyses Reveal Selection Signatures Underlying Adaptation and Production in Tibetan and Poll Dorset Sheep. Sci. Rep. 2021, 11, 2466. [Google Scholar] [CrossRef]
- Álvarez, I.; Fernández, I.; Traoré, A.; Pérez-Pardal, L.; Menéndez-Arias, N.A.; Goyache, F. Genomic Scan of Selective Sweeps in Djallonké (West African Dwarf) Sheep Shed Light on Adaptation to Harsh Environments. Sci. Rep. 2020, 10, 2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eydivandi, S.; Roudbar, M.A.; Ardestani, S.S.; Momen, M.; Sahana, G. A Selection Signatures Study among Middle Eastern and European Sheep Breeds. J. Anim. Breed. Genet. 2021, 138, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Gu, Z.; Guang, X.; Marín, J.C.; Varas, V.; González, B.A.; Wheeler, J.C.; Hu, Y.; Li, E.; Sun, X.; et al. Genomic Analysis of the Domestication and Post-Spanish Conquest Evolution of the Llama and Alpaca. Genome Biol. 2020, 21, 159. [Google Scholar] [CrossRef]
- Macciotta, N.P.P.; Colli, L.; Cesarani, A.; Ajmone-Marsan, P.; Low, W.Y.; Tearle, R.; Williams, J.L. The Distribution of Runs of Homozygosity in the Genome of River and Swamp Buffaloes Reveals a History of Adaptation, Migration and Crossbred Events. Genet. Sel. Evol. 2021, 53, 20. [Google Scholar] [CrossRef]
- Abied, A.; Xu, L.; Sahlu, B.W.; Xing, F.; Ahbara, A.; Pu, Y.; Lin, J.; Berihulay, H.; Islam, R.; He, X.; et al. Genome-Wide Analysis Revealed Homozygosity and Demographic History of Five Chinese Sheep Breeds Adapted to Different Environments. Genes 2020, 11, 1480. [Google Scholar] [CrossRef]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 595, 585–595. [Google Scholar] [CrossRef]
- Simonsen, K.L.; Churchill, G.A.; Aquadro, C.F. Properties of Statistical Tests of Neutrality for DNA Polymorphism Data. Genetics 1995, 141, 413–429. [Google Scholar] [CrossRef]
- Fay, J.C.; Wu, C.I. Hitchhiking under Positive Darwinian Selection. Genetics 2000, 155, 1405–1413. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, K.A.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B.P. Selection Signatures in Livestock Genome: A Review of Concepts, Approaches and Applications. Livest. Sci. 2020, 241, 104257. [Google Scholar] [CrossRef]
- Fariello, M.I.; Boitard, S.; Naya, H.; SanCristobal, M.; Servin, B. Detecting Signatures of Selection through Haplotype Differentiation among Hierarchically Structured Populations. Genetics 2013, 193, 929–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Neilsen, R. Linkage Disequilibrium as a Signature of Selective Sweeps. Genetics 2004, 167, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Sabeti, P.C.; Reich, D.E.; Higgins, J.M.; Levine, H.Z.P.; Richter, D.J.; Schaffner, S.F.; Gabriel, S.B.; Platko, J.V.; Patterson, N.J.; McDonald, G.J.; et al. Detecting Recent Positive Selection in the Human Genome from Haplotype Structure. Nature 2002, 419, 832–837. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-Wide Detection and Characterization of Positive Selection in Human Populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A Map of Recent Positive Selection in the Human Genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Choudhry, S.; Mei, R.; Morgan, M.; Rodriguez-Cintron, W.; Burchard, E.G.; Risch, N.J. Recent Genetic Selection in the Ancestral Admixture of Puerto Ricans. Am. J. Hum. Genet. 2007, 81, 626–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, J.; Morton, N.E.; Collins, A. Extended Tracts of Homozygosity in Outbred Human Populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisbin, A.; Bryc, K.; Byrnes, J.; Zakharia, F.; Omberg, L.; Degenhardt, J.; Reynolds, A.; Ostrer, H.; Mezey, J.G.; Bustamante, C.D. PCAdmix: Principal Components-Based Assignment of Ancestry along Each Chromosome in Individuals with Admixed Ancestry from Two or More Populations. Hum. Biol. 2012, 84, 343–364. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, E.G.; Maihofer, A.X.; Kanai, M.; Martin, A.R.; Karczewski, K.J.; Santoro, M.L.; Ulirsch, J.C.; Kamatani, Y.; Okada, Y.; Finucane, H.K.; et al. Tractor Uses Local Ancestry to Enable the Inclusion of Admixed Individuals in GWAS and to Boost Power. Nat. Genet. 2021, 53, 195–204. [Google Scholar] [CrossRef]
- Pasaniuc, B.; Sankararaman, S.; Kimmel, G.; Halperin, E. Inference of Locus-Specific Ancestry in Closely Related Populations. Bioinformatics 2009, 25, i213–i221. [Google Scholar] [CrossRef] [Green Version]
- Schubert, R.; Andaleon, A.; Wheeler, H.E. Comparing Local Ancestry Inference Models in Populations of Two- And Three-Way Admixture. PeerJ 2020, 8, e10090. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Y.; Zhao, Y. Systematic Review on Local Ancestor Inference From a Mathematical and Algorithmic Perspective. Front. Genet. 2021, 12, 639877. [Google Scholar] [CrossRef]
- Tang, H.; Coram, M.; Wang, P.; Zhu, X.; Risch, N. Reconstructing Genetic Ancestry Blocks in Admixed Individuals. Am. J. Hum. Genet. 2006, 79, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y. Detecting Structure of Haplotypes and Local Ancestry. Genetics 2014, 196, 625–642. [Google Scholar] [CrossRef] [Green Version]
- Salter-Townshend, M.; Myers, S. Fine-Scale Inference of Ancestry Segments without Prior Knowledge of Admixing Groups. Genetics 2019, 212, 869–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Dias-Alves, T.; Mairal, J.; Blum, M.G.B. Loter: A Software Package to Infer Local Ancestry for a Wide Range of Species. Mol. Biol. Evol. 2018, 35, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Utsunomiya, Y.T.; Milanesi, M.; Barbato, M.; Utsunomiya, A.T.H.; Sölkner, J.; Ajmone-Marsan, P.; Garcia, J.F. Unsupervised Detection of Ancestry Tracks with the GHap R Package. Methods Ecol. Evol. 2020, 11, 1448–1454. [Google Scholar] [CrossRef]
- Popescu, A.-A.; Huber, K.T. PSIKO2: A Fast and Versatile Tool to Infer Population Stratification on Various Levels in GWAS. Bioinformatics 2015, 31, 3552–3554. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Ma, Y.; Chen, A.; Fu, L.; Zhang, Y. Robust Sliding Mode Control for Markovian Jump Singular Systems with Randomly Changing Structure. Appl. Math. Comput. 2019, 349, 81–96. [Google Scholar] [CrossRef]
- Barbato, M.; Hailer, F.; Orozco-terWengel, P.; Kijas, J.W.; Mereu, P.; Cabras, P.; Mazza, R.; Pirastru, M.; Bruford, M.W. Genomic Signatures of Adaptive Introgression from European Mouflon into Domestic Sheep. Sci. Rep. 2017, 7, 7623. [Google Scholar] [CrossRef] [Green Version]
- Barbato, M.; Hailer, F.; Upadhyay, M.; Del Corvo, M.; Colli, L.; Negrini, R.; Kim, E.; Crooijmans, R.P.M.A.; Sonstegard, T.; Ajmone-marsan, P. Adaptive Introgression from Indicine Cattle into White Cattle Breeds from Central Italy. Sci. Rep. 2020, 10, 1279. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ding, X.-D.; Wang, S.; Wójcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.-S.; Faruque, O.; Nielsen, R.; et al. Pervasive Introgression Facilitated Domestication and Adaptation in the Bos Species Complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef]
- Hu, X.-J.; Yang, J.; Xie, X.-L.; Lv, F.-H.; Cao, Y.-H.; Li, W.-R.; Liu, M.-J.; Wang, Y.-T.; Li, J.-Q.; Liu, Y.-G.; et al. The Genome Landscape of Tibetan Sheep Reveals Adaptive Introgression from Argali and the History of Early Human Settlements on the Qinghai–Tibetan Plateau. Mol. Biol. Evol. 2019, 36, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kwon, T.; Dessie, T.; Yoo, D.; Mwai, O.A.; Jang, J.; Sung, S.; Lee, S.; Salim, B.; Jung, J.; et al. The Mosaic Genome of Indigenous African Cattle as a Unique Genetic Resource for African Pastoralism. Nat. Genet. 2020, 52, 1099–1110. [Google Scholar] [CrossRef]
- Manel, S.; Schwartz, M.K.; Luikart, G.; Taberlet, P. Landscape Genetics: Combining Landscape Ecology and Population Genetics. Trends Ecol. Evol. 2003, 18, 189–197. [Google Scholar] [CrossRef]
- Jelinski, D.E. On Genes and Geography: A Landscape Perspective on Genetic Variation in Natural Plant Populations. Landsc. Urban Plan. 1997, 39, 11–23. [Google Scholar] [CrossRef]
- Goodchild, M. Geographical Information Science. Int. J. Geogr. Inf. Syst. 1992, 6, 31–45. [Google Scholar] [CrossRef]
- Joost, S.; Bonin, A.; Bruford, M.; Despres, L.; Concord, C.; Erhardt, G.; Taberlet, P. A Spatial Analysis Method (SAM) to Detect Candidate Loci for Selection: Towards a Landscape Genomics Approach to Adaptation. Mol. Ecol. 2007, 16, 3955–3969. [Google Scholar] [CrossRef]
- Joost, S.; Kalbermatten, M.; Bonin, A. Spatial Analysis Method (Sam): A Software Tool Combining Molecular and Environmental Data to Identify Candidate Loci for Selection. Mol. Ecol. Resour. 2008, 8, 957–960. [Google Scholar] [CrossRef]
- Pariset, L.; Joost, S.; Ajmone Marsan, P.; Valentini, A.; Econogene Consortium (EC). Landscape Genomics and Biased F ST Approaches Reveal Single Nucleotide Polymorphisms under Selection in Goat Breeds of North-East Mediterranean. BMC Genet. 2009, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Tonteri, A.; Vasemagi, A.; Lumme, J.; Primmer, C.R. Beyond MHC: Signals of Elevated Selection Pressure on Atlantic Salmon (Salmo salar) Immune-Relevant Loci. Mol. Ecol. 2010, 19, 1273–1282. [Google Scholar] [CrossRef]
- Frichot, E.; Schoville, S.D.; Bouchard, G.; Franc, O. Testing for Associations between Loci and Environmental Gradients Using Latent Factor Mixed Models. Mol. Biol. Evol. 2013, 30, 1687–1699. [Google Scholar] [CrossRef] [Green Version]
- Mdladla, K.; Dzomba, E.F. Landscape Genomics and Pathway Analysis to Understand Genetic Adaptation of South African Indigenous Goat Populations. Heredity (Edinb) 2018, 120, 369–378. [Google Scholar] [CrossRef]
- Stucki, S.; Orozco-Terwengel, P.; Forester, B.R.; Duruz, S.; Colli, L.; Masembe, C. High Performance Computation of Landscape Genomic Models Including Local Indicators of Spatial Association. Mol. Ecol. Resour. 2017, 17, 1072–1089. [Google Scholar] [CrossRef] [Green Version]
- Cortellari, M.; Barbato, M.; Talenti, A.; Bionda, A.; Randi, E.; Sarti, F.M.; Sartore, S.; Soglia, D.; Liotta, L. The Climatic and Genetic Heritage of Italian Goat Breeds with Genomic SNP Data. Sci. Rep. 2021, 11, 10986. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Yang, H.; Wang, S.; Rong, E.; Pei, W.; Li, H.; Wang, N. Genome-Wide Association Study for Wool Production Traits in a Chinese Merino Sheep Population. PLoS ONE 2014, 9, e107101. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.B.S.; Fonseca, L.F.S.; Pinheiro, D.G.; Magalhães, A.F.B.; Muniz, M.M.M.; Ferro, J.A.; Baldi, F.; Chardulo, L.A.L.; Schnabel, R.D.; Taylor, J.F.; et al. Spliced Genes in Muscle from Nelore Cattle and Their Association with Carcass and Meat Quality. Sci. Rep. 2020, 10, 14701. [Google Scholar] [CrossRef]
- Colli, L.; Negrini, R.; Nicoloso, L.; Crepaldi, P. Assessing The Spatial Dependence of Adaptive Loci in 43 European and Western Asian Goat Breeds Using AFLP Markers. PLoS ONE 2014, 9, e86668. [Google Scholar] [CrossRef] [PubMed]
- Buitkamp, J.; Filmether, P.; Stear, M.; Epplen, J. Class I and Class II Major Histocompatibility Complex Alleles Are Associated with Faecal Egg Counts Following Natural, Predominantly Ostertagia Circumcincta Infection. Parasitol. Res. 1996, 82, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Duruz, S.; Rochat, E.; Selmoni, O.; Vajana, E.; Orozco, P.; Joost, S.; Dunner, S.; Bruford, C.M.W. Rapid Identification and Interpretation of Gene—Environment Associations Using the New R. SamBada Landscape Genomics Pipeline. Mol. Ecol. Resour. 2019, 19, 1355–1365. [Google Scholar] [CrossRef] [Green Version]
- Gunther, T.; Coop, G. Robust Identification of Local Adaptation from Allele Frequencies. Genetics 2013, 195, 205–220. [Google Scholar] [CrossRef] [Green Version]
- Frichot, E.; Francois, O. LEA: An R Package for Landscape and Ecological Association Studies. Methods Ecol. Evol. 2015, 6, 925–929. [Google Scholar] [CrossRef]
- Guillot, G.; Vitalis, R. Detecting Correlation between Allele Frequencies and Environmental Variables as a Signature of Selection. A Fast Computational Approach for Genome-Wide Studies. Spat. Stat. 2014, 8, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Coop, G.; Witonsky, D.; Di Rienzo, A.; Pritchard, J.K. Using Environmental Correlations to Identify Loci Underlying Local Adaptation. Genetics 2010, 185, 1411–1423. [Google Scholar] [CrossRef] [Green Version]
- Gautier, M. Genome-Wide Scan for Adaptive Divergence and Association with Population-Specific Covariates. Genetics 2015, 201, 1555–1579. [Google Scholar] [CrossRef] [Green Version]
- De Villemereuil, P.; Gaggiotti, O.E. A New F ST-Based Method to Uncover Local Adaptation Using Environmental Variables. Methods Ecol. Evol. 2015, 6, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Caye, K.; Jumentier, B.; Lepeule, J.; François, O. LFMM 2: Fast and Accurate Inference of Gene-Environment Associations in Genome-Wide Studies. Mol. Biol. Evol. 2019, 36, 852–860. [Google Scholar] [CrossRef]
- Antao, T.; Lopes, A.; Lopes, R.J.; Beja-pereira, A.; Luikart, G. LOSITAN: A Workbench to Detect Molecular Adaptation Based on a F St -Outlier Method. BMC Bioinform. 2008, 5, 323. [Google Scholar] [CrossRef] [Green Version]
- Beaumont, M.A.; Nichols, R.A. Evaluating Loci for Use in the Genetic Analysis of Population Structure. Proc. R. Soc. Lond. B. 1996, 263, 1619–1623. [Google Scholar]
- Foll, M.; Gaggiotti, O. A Genome-Scan Method to Identify Selected Loci Appropriate for Both Dominant and Codominant Markers: A Bayesian Perspective. Genetics 2008, 993, 977–993. [Google Scholar] [CrossRef] [Green Version]
- Landguth, E.; Cushman, S.; Schwartz, M.; McKelvey, K.; Murphy, M.; Luikart, G. Quantifying the Lag Time to Detect Barriers in Landscape Genetics. Mol. Ecol. 2010, 19, 4179–4191. [Google Scholar] [CrossRef] [PubMed]
- De Mita, S.; Thuillet, A.; Gay, L.; Ahmadi, N.; Manel, S.; Ronfort, J. Detecting Selection along Environmental Gradients: Analysis of Eight Methods and Their Effectiveness for Outbreeding and Selfing Populations. Mol. Ecol. 2013, 22, 1383–1399. [Google Scholar] [CrossRef] [PubMed]
- Terasaki Hart, D.E.; Bishop, A.P.; Wang, I.J. Geonomics: Forward-Time, Spatially Explicit, and Arbitrarily Complex Landscape Genomic Simulations. Mol. Biol. Evol. 2021. [Google Scholar] [CrossRef]
- Stephens, Z.D.; Lee, S.Y.; Faghri, F.; Campbell, R.H.; Zhai, C.; Efron, M.J.; Iyer, R.; Schatz, M.C.; Sinha, S.; Robinson, G.E. Big Data: Astronomical or Genomical? PLoS Biol. 2015, 13, e1002195. [Google Scholar] [CrossRef]
- Neethirajan, S. Sensing and Bio-Sensing Research The Role of Sensors, Big Data and Machine Learning in Modern Animal Farming. Sens. Bio-Sens. Res. 2020, 29, 100367. [Google Scholar] [CrossRef]
- Rees, J.S.; Castellano, S.; Andrés, A.M. The Genomics of Human Local Adaptation. Trends Genet. 2020, 36, 415–428. [Google Scholar] [CrossRef]
- Schrider, D.R.; Kern, A.D. Soft Sweeps Are the Dominant Mode of Adaptation in the Human Genome. Mol. Biol. Evol. 2017, 34, 1863–1877. [Google Scholar] [CrossRef] [Green Version]
- Sugden, L.A.; Atkinson, E.G.; Fischer, A.P.; Rong, S.; Henn, B.M.; Ramachandran, S. Localization of Adaptive Variants in Human Genomes Using Averaged One-Dependence Estimation. Nat. Commun. 2018, 9, 703. [Google Scholar] [CrossRef] [Green Version]
- Nayeri, S.; Sargolzaei, M.; Tulpan, D. A Review of Traditional and Machine Learning Methods Applied to Animal Breeding. Anim. Health Res. Rev. 2019, 20, 31–46. [Google Scholar] [CrossRef]
- Okser, S.; Pahikkala, T.; Airola, A.; Salakoski, T.; Ripatti, S.; Aittokallio, T. Regularized Machine Learning in the Genetic Prediction of Complex Traits. PLoS Genet. 2014, 10, e1004754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arpanahi Abdollahi, R.; Gianola, D.; Peñagaricano, F. Deep Learning versus Parametric and Ensemble Methods for Genomic Prediction of Complex Phenotypes. Genet. Sel. Evol. 2020, 52, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhang, N.; Wang, Y.-G.; George, A.W.; Reverter, A.; Li, Y. Genomic Prediction of Breeding Values Using a Subset of SNPs Identified by Three Machine Learning Methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef]
- Waldmann, P. Approximate Bayesian Neural Networks in Genomic Prediction. Genet. Sel. Evol. 2018, 50, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, P.; Campos, G.D.L.; Pérez-enciso, M. Can Deep Learning Improve Genomic Prediction of Complex Human Traits? Genetics 2018, 210, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Piles, M.; Lozano, C.F.; Galilea, M.V.; Rodríguez, O.G.; Sánchez, J.P.; Torrallardona, D.; Ballester, M.; Quintanilla, R. Machine Learning Applied to Transcriptomic Data to Identify Genes Associated with Feed Efficiency in Pigs. Genet. Sel. Evol. 2019, 51, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaluarachchi, T.; Reis, A.; Nanayakkara, S. A Review of Recent Deep Learning Approaches in Human-Centered Machine Learning. Sensors 2021, 21, 2514. [Google Scholar] [CrossRef] [PubMed]
- Helm, J.M.; Swiergosz, A.M.; Haeberle, H.S.; Karnuta, J.M.; Schaffer, J.L.; Krebs, V.E.; Spitzer, A.I.; Ramkumar, P.N. Machine Learning and Artificial Intelligence: Definitions, Applications, and Future Directions. Curr. Rev. Musculoskelet. Med. 2020, 13, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.I.; Mitchell, T.M. Machine Learning: Trends, Perspectives, and Prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Schrider, D.R.; Kern, A.D. Supervised Machine Learning for Population Genetics: A New Paradigm. Trends Genet. 2018, 34, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Harfouche, A.L.; Jacobson, D.A.; Kainer, D.; Romero, J.C.; Harfouche, A.H.; Scarascia Mugnozza, G.; Moshelion, M.; Tuskan, G.A.; Keurentjes, J.J.B.; Altman, A. Accelerating Climate Resilient Plant Breeding by Applying Next-Generation Artificial Intelligence. Trends Biotechnol. 2019, 37, 1217–1235. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep Learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, D.; He, F.; Wang, J.; Joshi, T.; Xu, D. Phenotype Prediction and Genome-Wide Association Study Using Deep Convolutional Neural Network of Soybean. Front. Genet. 2019, 10, 1091. [Google Scholar] [CrossRef]
- Voosen, P. How AI Detectives Are Cracking Open the Black Box of Deep Learning. Science 2017. [Google Scholar] [CrossRef]
- Hu, T.; Darabos, C.; Urbanowicz, R. Editorial: Machine Learning in Genome-Wide Association Studies. Front. Genet. 2020, 11, 593958. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, as- Sociation Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Maclean, C.A.; Hong, N.P.C.; Prendergast, J.G.D. Hapbin: An Efficient Program for Performing Haplotype-Based Scans for Positive Selection in Large Genomic Datasets. Mol. Biol. Evol. 2015, 32, 3027–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudet, J. HIERFSTAT, a Package for R to Compute and Test Hierarchical F-Statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W. Robust Relationship Inference in Genome-Wide Association Studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, B.; Wittelsbu, U.; Ramos-onsins, S.E.; Lercher, M.J. PopGenome: An Efficient Swiss Army Knife for Population Genomic Analyses in R. Mol. Biol. Evol. 2014, 31, 1929–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofler, R.; Orozco-terwengel, P.; De Maio, N.; Pandey, R.V.; Nolte, V.; Kosiol, C.; Schlo, C. PoPoolation: A Toolbox for Population Genetic Analysis of Next Generation Sequencing Data from Pooled Individuals. PLoS ONE 2011, 6, e15925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, M.; Klassmann, A.; Vitalis, R. REHH 2.0: A Reimplementation of the R Package REHH to Detect Positive Selection from Haplotype Structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An Efficient Multithreaded Program to Perform EHH-Based Scans for Positive Selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [Green Version]
- Vilella, A.J.; Blanco-Garcia, A.; Hutter, S.; Rozas, J. VariScan: Analysis of Evolutionary Patterns from Large-Scale DNA Sequence Polymorphism Data. Bioinformatics 2005, 21, 2791–2793. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; Depristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.-Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance Component Model to Account for Sample Structure in Genome-Wide Association Studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, G.; Lee, S.H.; Hayes, B.J.; Goddard, M.E.; Wray, N.R.; Visscher, P.M. Simultaneous Discovery, Estimation and Prediction Analysis of Complex Traits Using a Bayesian Mixture Model. PLoS ONE 2015, 11, e1004969. [Google Scholar] [CrossRef] [PubMed]
- do Prado Paim, T.; Hay, E.H.A.; Wilson, C.; Thomas, M.G.; Kuehn, L.A.; Paiva, S.R.; Mcmanus, C.; Blackburn, H. Genomic Breed Composition of Selection Signatures in Brangus Beef Cattle. Front. Genet. 2020, 11, 710. [Google Scholar] [CrossRef]
- Francis, J.; Little, D.A. Resistance of Droughtmaster Cattle to Tick Infestation and Babesiosis. Aust. Vet. J. 1964, 40, 247–253. [Google Scholar] [CrossRef]
- O’Neill, C.; Swain, D.; Kadarmideen, H. Evolutionary Process of Bos Taurus Cattle in Favourable versus Unfavourable Environments and Its Implications for Genetic Selection. Evol. Appl. 2010, 3, 422–433. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Passamonti, M.M.; Somenzi, E.; Barbato, M.; Chillemi, G.; Colli, L.; Joost, S.; Milanesi, M.; Negrini, R.; Santini, M.; Vajana, E.; et al. The Quest for Genes Involved in Adaptation to Climate Change in Ruminant Livestock. Animals 2021, 11, 2833. https://doi.org/10.3390/ani11102833
Passamonti MM, Somenzi E, Barbato M, Chillemi G, Colli L, Joost S, Milanesi M, Negrini R, Santini M, Vajana E, et al. The Quest for Genes Involved in Adaptation to Climate Change in Ruminant Livestock. Animals. 2021; 11(10):2833. https://doi.org/10.3390/ani11102833
Chicago/Turabian StylePassamonti, Matilde Maria, Elisa Somenzi, Mario Barbato, Giovanni Chillemi, Licia Colli, Stéphane Joost, Marco Milanesi, Riccardo Negrini, Monia Santini, Elia Vajana, and et al. 2021. "The Quest for Genes Involved in Adaptation to Climate Change in Ruminant Livestock" Animals 11, no. 10: 2833. https://doi.org/10.3390/ani11102833