Levelling the Translational Gap for Animal to Human Efficacy Data

 ,
,
Simple Summary
Abstract
1. Introduction
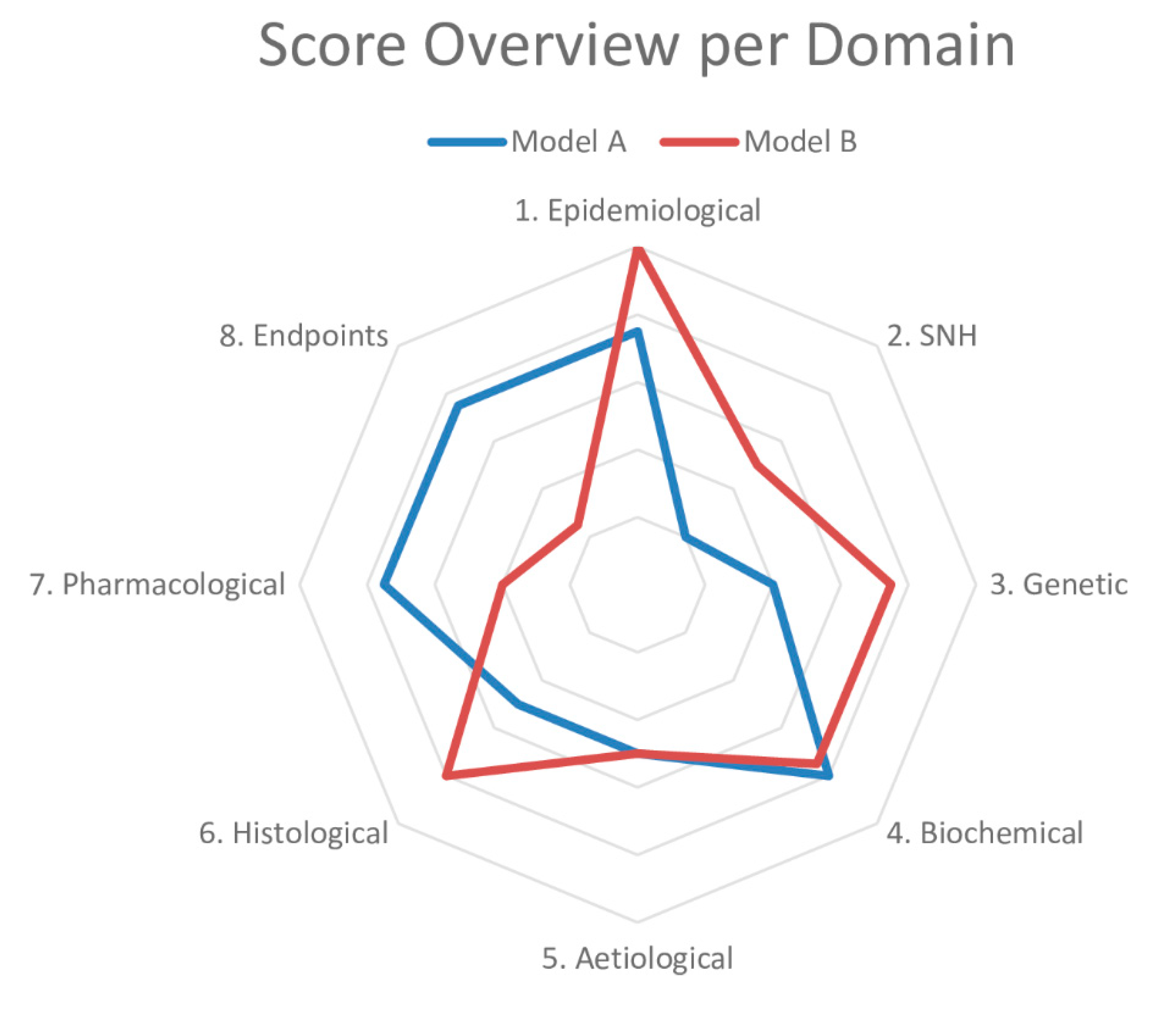
2. The Framework to Identify Models of Disease (FIMD)
3. Systematic Review and Meta-Analysis
4. Can Animal Models Predict Human Pharmacologically Active Ranges? A First Glance into the Investigator’s Brochure
5. Levelling the Translational Gap for Animal to Human Efficacy Data
6. Final Considerations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Pammolli, F.; Righetto, L.; Abrignani, S.; Pani, L.; Pelicci, P.G.; Rabosio, E. The endless frontier? The recent increase of R&D productivity in pharmaceuticals. J. Transl. Med. 2020, 18, 162. [Google Scholar] [CrossRef] [PubMed]
- Van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef]
- Schulz, J.B.; Cookson, M.R.; Hausmann, L. The impact of fraudulent and irreproducible data to the translational research crisis—Solutions and implementation. J. Neurochem. 2016, 139, 253–270. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. Acknowledging and overcoming nonreproducibility in basic and preclinical research. JAMA 2017, 317, 1019. [Google Scholar] [CrossRef]
- Vogt, L.; Reichlin, T.S.; Nathues, C.; Würbel, H. Authorization of animal experiments is based on confidence rather than evidence of scientific rigor. PLoS Biol. 2016, 14, e2000598. [Google Scholar] [CrossRef]
- Begley, C.G.; Ellis, L.M. Drug development: Raise standards for preclinical cancer research. Nature 2012, 483, 531–533. [Google Scholar] [CrossRef]
- Prinz, F.; Schlange, T.; Asadullah, K. Believe it or not: How much can we rely on published data on potential drug targets? Nat. Rev. Drug Discov. 2011, 10, 712. [Google Scholar] [CrossRef]
- Perrin, S. Preclinical research: Make mouse studies work. Nature 2014, 507, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef] [PubMed]
- Bebarta, V.; Luyten, D.; Heard, K. Emergency medicine animal research: Does use of randomization and blinding affect the results? Acad. Emerg. Med. 2003, 10, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Pogoda, A.; Bonberg, N.; Koecke, M.H.M.; Strecker, J.; Wellmann, J.; Bruckmann, N.; Beuker, C.; Schäbitz, W.; Meuth, S.G.; Wiendl, H.; et al. Why most acute stroke studies are positive in animals but not in patients: A systematic comparison of preclinical, early phase, and phase 3 clinical trials of neuroprotective agents. Ann. Neurol. 2020, 87, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Clutton, R.E.; Lilley, E.; Hansen, K.E.A.; Brattelid, T. PREPARE: Guidelines for planning animal research and testing. Lab. Anim. 2018, 52, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.; Avey, M.T.; Anestidou, L.; Ritskes-Hoitinga, M.; Griffin, G. Improving animal research reporting standards: HARRP, the first step of a unified approach by ICLAS to improve animal research reporting standards worldwide. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Baker, D.; Lidster, K.; Sottomayor, A.; Amor, S. Two years later: Journals are not yet enforcing the ARRIVE guidelines on reporting standards for pre-clinical animal studies. PLoS Biol. 2014, 12, e1001756. [Google Scholar] [CrossRef]
- Henderson, V.C.; Kimmelman, J.; Fergusson, D.; Grimshaw, J.M.; Hackam, D.G. Threats to validity in the design and conduct of preclinical efficacy studies: A systematic review of guidelines for in vivo animal experiments. PLoS Med. 2013, 10, e1001489. [Google Scholar] [CrossRef]
- Hackam, D.G. Translating animal research into clinical benefit. BMJ 2007, 334, 163–164. [Google Scholar] [CrossRef]
- Sams-Dodd, F. Strategies to optimize the validity of disease models in the drug discovery process. Drug Discov. Today 2006, 11, 355–363. [Google Scholar] [CrossRef]
- Ferreira, S.G.; Veening-Griffioen, D.H.; Boon, W.P.C.; Moors, E.H.M.; Gispen-de Wied, C.C.; Schellekens, H.; van Meer, P.J.K. A standardised framework to identify optimal animal models for efficacy assessment in drug development. PLoS ONE 2019, 14, e0218014. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.C.; Moors, E.H.M.; Gispen-de Wied, C.C.; Schellekens, H.; van Meer, P.J.K. Correction: A standardised framework to identify optimal animal models for efficacy assessment in drug development. PLoS ONE 2019, 14, e0220325. [Google Scholar] [CrossRef] [PubMed]
- Macleod, M.R.; Lawson McLean, A.; Kyriakopoulou, A.; Serghiou, S.; de Wilde, A.; Sherratt, N.; Hirst, T.; Hemblade, R.; Bahor, Z.; Nunes-Fonseca, C.; et al. Risk of bias in reports of in vivo research: A focus for improvement. PLoS Biol. 2015, 13, e1002273. [Google Scholar] [CrossRef] [PubMed]
- Langhof, H.; Chin, W.W.L.; Wieschowski, S.; Federico, C.; Kimmelman, J.; Strech, D. Preclinical efficacy in therapeutic area guidelines from the U.S. Food and Drug Administration and the European Medicines Agency: A cross-sectional study. Br. J. Pharmacol. 2018, 175, 4229–4238. [Google Scholar] [CrossRef]
- Varga, O.E.; Hansen, A.K.; Sandøe, P.; Olsson, I.A.S. Validating animal models for preclinical research: A scientific and ethical discussion. Altern. Lab. Anim. 2010, 38, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Wieschowski, S.; Chin, W.W.L.; Federico, C.; Sievers, S.; Kimmelman, J.; Strech, D. Preclinical efficacy studies in investigator brochures: Do they enable risk–benefit assessment? PLoS Biol. 2018, 16, e2004879. [Google Scholar] [CrossRef] [PubMed]
- McKinney, W.T. Animal model of depression: I. Review of evidence: Implications for research. Arch. Gen. Psychiatry 1969, 21, 240. [Google Scholar] [CrossRef]
- Willner, P. The validity of animal models of depression. Psychopharmacology 1984, 83, 1–16. [Google Scholar] [CrossRef]
- Denayer, T.; Stöhr, T.; Roy, M.V. Animal models in translational medicine: Validation and prediction. Eur. J. Mol. Clin. Med. 2014, 2, 5. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Rovers, M.M.; de Vries, R.B.M.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef]
- Yu, X.; Bao, B.; Echigoya, Y.; Yokota, T. Dystrophin-deficient large animal models: Translational research and exon skipping. Am. J. Transl. Res. 2015, 7, 1314–1331. [Google Scholar] [PubMed]
- Pound, P.; Ebrahim, S.; Sandercock, P.; Bracken, M.B.; Roberts, I. Where is the evidence that animal research benefits humans? BMJ 2004, 328, 514–517. [Google Scholar] [CrossRef]
- Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.C.; Hooijmans, C.R.; Moors, E.H.M.; Schellekens, H.; van Meer, P.J.K. Comparison of drug efficacy in two animal models of type 2 diabetes: A systematic review and meta-analysis. Eur. J. Pharmacol. 2020, 879, 173153. [Google Scholar] [CrossRef] [PubMed]
- FDA. Guidance for Industry Diabetes Mellitus: Developing Drugs and Therapeutic Biologics for Treatment and Prevention; FDA: Washington, DC, USA, 2008. [Google Scholar]
- EMA. Guideline on Clinical Investigation of Medicinal Products in 5 the Treatment or Prevention of Diabetes Mellitus; FDA: Washington, DC, USA, 2018. [Google Scholar]
- Leenaars, C.H.C.; Kouwenaar, C.; Stafleu, F.R.; Bleich, A.; Ritskes-Hoitinga, M.; De Vries, R.B.M.; Meijboom, F.L.B. Animal to human translation: A systematic scoping review of reported concordance rates. J. Transl. Med. 2019, 17, 223. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH E6 (R2) Good Clinical Practice—Step 5; European Medicines Agency: London, UK, 2016.
- Van Gerven, J.; Cohen, A. Integrating data from the investigational medicinal product dossier/investigator’s brochure. A new tool for translational integration of preclinical effects. Br. J. Clin. Pharmacol. 2018, 84, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J. Improving the predictive value of interventional animal models data. Drug Discov. Today 2015, 20, 475–482. [Google Scholar] [CrossRef]
- Clark, M.; Steger-Hartmann, T. A big data approach to the concordance of the toxicity of pharmaceuticals in animals and humans. Regul. Toxicol. Pharmacol. 2018, 96, 94–105. [Google Scholar] [CrossRef]
- Veening-Griffioen, D.H.; Ferreira, G.S.; van Meer, P.J.K.; Boon, W.P.C.; Gispen-de Wied, C.C.; Moors, E.H.M.; Schellekens, H. Are some animal models more equal than others? A case study on the translational value of animal models of efficacy for Alzheimer’s disease. Eur. J. Pharmacol. 2019, 859, 172524. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef]
- Freedman, L.P.; Cockburn, I.M.; Simcoe, T.S. The economics of reproducibility in preclinical research. PLoS Biol. 2015, 13, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Bannach-Brown, A.; Przybyła, P.; Thomas, J.; Rice, A.S.C.; Ananiadou, S.; Liao, J.; Macleod, M.R. Machine learning algorithms for systematic review: Reducing workload in a preclinical review of animal studies and reducing human screening error. Syst. Rev. 2019, 8, 23. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Shin, D.; Vander Wyk, B.; Beck, A.P.; Zatz, N.; Sneiderman, C.A.; Kilicoglu, H. Menagerie: A text-mining tool to support animal-human translation in neurodegeneration research. PLoS ONE 2019, 14, e0226176. [Google Scholar] [CrossRef]
- Veening-Griffioen, D.H.; Ferreira, G.S.; Boon, W.P.C.; Gispen-de Wied, C.C.; Schellekens, H.; Moors, E.H.M.; van Meer, P.J.K. Tradition, not science, is the basis of animal model selection in translational and applied research. ALTEX 2020. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, J.; Henderson, V. Assessing risk/benefit for trials using preclinical evidence: A proposal. J. Med. Ethics 2016, 42, 50–53. [Google Scholar] [CrossRef]
- Hair, K.; Macleod, M.R.; Sena, E.S. A randomised controlled trial of an Intervention to Improve Compliance with the ARRIVE guidelines (IICARus). Res. Integr. Peer Rev. 2019, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Avila, A.M.; Bebenek, I.; Bonzo, J.A.; Bourcier, T.; Davis Bruno, K.L.; Carlson, D.B.; Dubinion, J.; Elayan, I.; Harrouk, W.; Lee, S.-L.; et al. An FDA/CDER perspective on nonclinical testing strategies: Classical toxicology approaches and new approach methodologies (NAMs). Regul. Toxicol. Pharmacol. 2020, 114, 104662. [Google Scholar] [CrossRef] [PubMed]
- Sheean, M.E.; Malikova, E.; Duarte, D.; Capovilla, G.; Fregonese, L.; Hofer, M.P.; Magrelli, A.; Mariz, S.; Mendez-Hermida, F.; Nistico, R.; et al. Nonclinical data supporting orphan medicinal product designations in the area of rare infectious diseases. Drug Discov. Today 2020, 25, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Sena, E.S.; Macleod, M.R. Bringing rigour to translational medicine. Nat. Rev. Neurol. 2014, 10, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ioannidis, J.P.A. Reproducibility in science: Improving the standard for basic and preclinical research. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, J.; Anderson, J.A. Should preclinical studies be registered? Nat. Biotechnol. 2012, 30, 488–489. [Google Scholar] [CrossRef]
- Preclinical Trials. PreclinicalTrials.eu. Available online: https://preclinicaltrials.eu/ (accessed on 20 May 2020).
- Viergever, R.F.; Karam, G.; Reis, A.; Ghersi, D. The quality of registration of clinical trials: Still a problem. PLoS ONE 2014, 9, e84727. [Google Scholar] [CrossRef] [PubMed]
- DeVito, N.J.; Bacon, S.; Goldacre, B. Compliance with legal requirement to report clinical trial results on ClinicalTrials.gov: A cohort study. Lancet 2020, 395, 361–369. [Google Scholar] [CrossRef]
- Van Meer, P.J.K.; Graham, M.L.; Schuurman, H.-J. The safety, efficacy and regulatory triangle in drug development: Impact for animal models and the use of animals. Eur. J. Pharmacol. 2015, 759, 3–13. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Handbook: Quality Practices in Basic Biomedical Research. Available online: https://www.who.int/tdr/publications/training-guideline-publications/handbook-quality-practices-biomedical-research/en/ (accessed on 23 May 2020).
- Ter Riet, G.; Korevaar, D.A.; Leenaars, M.; Sterk, P.J.; Van Noorden, C.J.F.; Bouter, L.M.; Lutter, R.; Elferink, R.P.O.; Hooft, L. Publication bias in laboratory animal research: A survey on magnitude, drivers, consequences and potential solutions. PLoS ONE 2012, 7, e43404. [Google Scholar] [CrossRef]
- Kimmelman, J.; Federico, C. Consider drug efficacy before first-in-human trials. Nature 2017, 542, 25–27. [Google Scholar] [CrossRef]
- Bailey, J.; Balls, M. Recent efforts to elucidate the scientific validity of animal-based drug tests by the pharmaceutical industry, pro-testing lobby groups, and animal welfare organisations. BMC Med. Ethics 2019, 20, 16. [Google Scholar] [CrossRef]
- Monticello, T.M.; Jones, T.W.; Dambach, D.M.; Potter, D.M.; Bolt, M.W.; Liu, M.; Keller, D.A.; Hart, T.K.; Kadambi, V.J. Current nonclinical testing paradigm enables safe entry to first-in-human clinical trials: The IQ consortium nonclinical to clinical translational database. Toxicol. Appl. Pharmacol. 2017, 334, 100–109. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials. JACC Basic Transl. Sci. 2020, 5, 387–397. [Google Scholar] [CrossRef]
- Haddrick, M.; Simpson, P.B. Organ-on-a-chip technology: Turning its potential for clinical benefit into reality. Drug Discov. Today 2019, 24, 1217–1223. [Google Scholar] [CrossRef]
- Vives, J.; Batlle-Morera, L. The challenge of developing human 3D organoids into medicines. Stem Cell Res. Ther. 2020, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.E.; Karp, J.M. All models are wrong, but some organoids may be useful. Genome Biol. 2019, 20, 66. [Google Scholar] [CrossRef] [PubMed]
- Hartung, T.; De Vries, R.; Hoffmann, S.; Hogberg, H.T.; Smirnova, L.; Tsaioun, K.; Whaley, P.; Leist, M. Toward good in vitro reporting standards. ALTEX 2019, 36, 3–17. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Weight | |
|---|---|
| 1. EPIDEMIOLOGICAL VALIDATION | 12.5 |
| 1.1 Is the model able to simulate the disease in the relevant sexes? | 6.25 |
| 1.2 Is the model able to simulate the disease in the relevant age groups (e.g., juvenile, adult or ageing)? | 6.25 |
| 2. SYMPTOMATOLOGY AND NATURAL HISTORY VALIDATION | 12.5 |
| 2.1 Is the model able to replicate the symptoms and co-morbidities commonly present in this disease? If so, which ones? | 2.5 |
| 2.2 Is the natural history of the disease similar to human’s regarding: 2.2.1 Time to onset | 2.5 |
| 2.2.2 Disease progression | 2.5 |
| 2.2.3 Duration of symptoms | 2.5 |
| 2.2.4 Severity | 2.5 |
| 3. GENETIC VALIDATION | 12.5 |
| 3.1 Does this species also have orthologous genes and/or proteins involved in the human disease? | 4.17 |
| 3.2 If so, are the relevant genetic mutations or alterations also present in the orthologous genes/proteins? | 4.17 |
| 3.3 If so, is the expression of such orthologous genes and/or proteins similar to the human condition? | 4.16 |
| 4. BIOCHEMICAL VALIDATION | 12.5 |
| 4.1 If there are known pharmacodynamic (PD) biomarkers related to the pathophysiology of the disease, are they also present in the model? | 3.125 |
| 4.2 Do these PD biomarkers behave similarly to humans’? | 3.125 |
| 4.3 If there are known prognostic biomarkers related to the pathophysiology of the disease, are they also present in the model? | 3.125 |
| 4.4 Do these prognostic biomarkers behave similarly to humans’? | 3.125 |
| 5. AETIOLOGICAL VALIDATION | 12.5 |
| 5.1 Is the aetiology of the disease similar to humans’? | 12.5 |
| 6. HISTOLOGICAL VALIDATION | 12.5 |
| 6.1 Do the histopathological structures in relevant tissues resemble the ones found in humans? | 12.5 |
| 7. PHARMACOLOGICAL VALIDATION | 12.5 |
| 7.1 Are effective drugs in humans also effective in this model? | 4.17 |
| 7.2 Are ineffective drugs in humans also ineffective in this model? | 4.17 |
| 7.3 Have drugs with different mechanisms of action and acting on different pathways been tested in this model? If so, which? | 4.16 |
| 8. ENDPOINT VALIDATION | 12.5 |
| 8.1 Are the endpoints used in preclinical studies the same or translatable to the clinical endpoints? | 6.25 |
| 8.2 Are the methods used to assess preclinical endpoints comparable to the ones used to assess related clinical endpoints? | 6.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, G.S.; Veening-Griffioen, D.H.; Boon, W.P.C.; Moors, E.H.M.; van Meer, P.J.K. Levelling the Translational Gap for Animal to Human Efficacy Data. Animals 2020, 10, 1199. https://doi.org/10.3390/ani10071199
Ferreira GS, Veening-Griffioen DH, Boon WPC, Moors EHM, van Meer PJK. Levelling the Translational Gap for Animal to Human Efficacy Data. Animals. 2020; 10(7):1199. https://doi.org/10.3390/ani10071199
Chicago/Turabian StyleFerreira, Guilherme S., Désirée H. Veening-Griffioen, Wouter P. C. Boon, Ellen H. M. Moors, and Peter J. K. van Meer. 2020. "Levelling the Translational Gap for Animal to Human Efficacy Data" Animals 10, no. 7: 1199. https://doi.org/10.3390/ani10071199
APA StyleFerreira, G. S., Veening-Griffioen, D. H., Boon, W. P. C., Moors, E. H. M., & van Meer, P. J. K. (2020). Levelling the Translational Gap for Animal to Human Efficacy Data. Animals, 10(7), 1199. https://doi.org/10.3390/ani10071199