Microbial Communities Associated with Farmed Genypterus chilensis: Detection in Water Prior to Bacterial Outbreaks Using Culturing and High-Throughput Sequencing


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Fish Maintenance Conditions
2.2. Seawater Sampling
2.3. Culturing Methods
2.4. Culture-Independent Methods
2.5. Diversity Analyses
3. Results
3.1. Bacterial Abundance
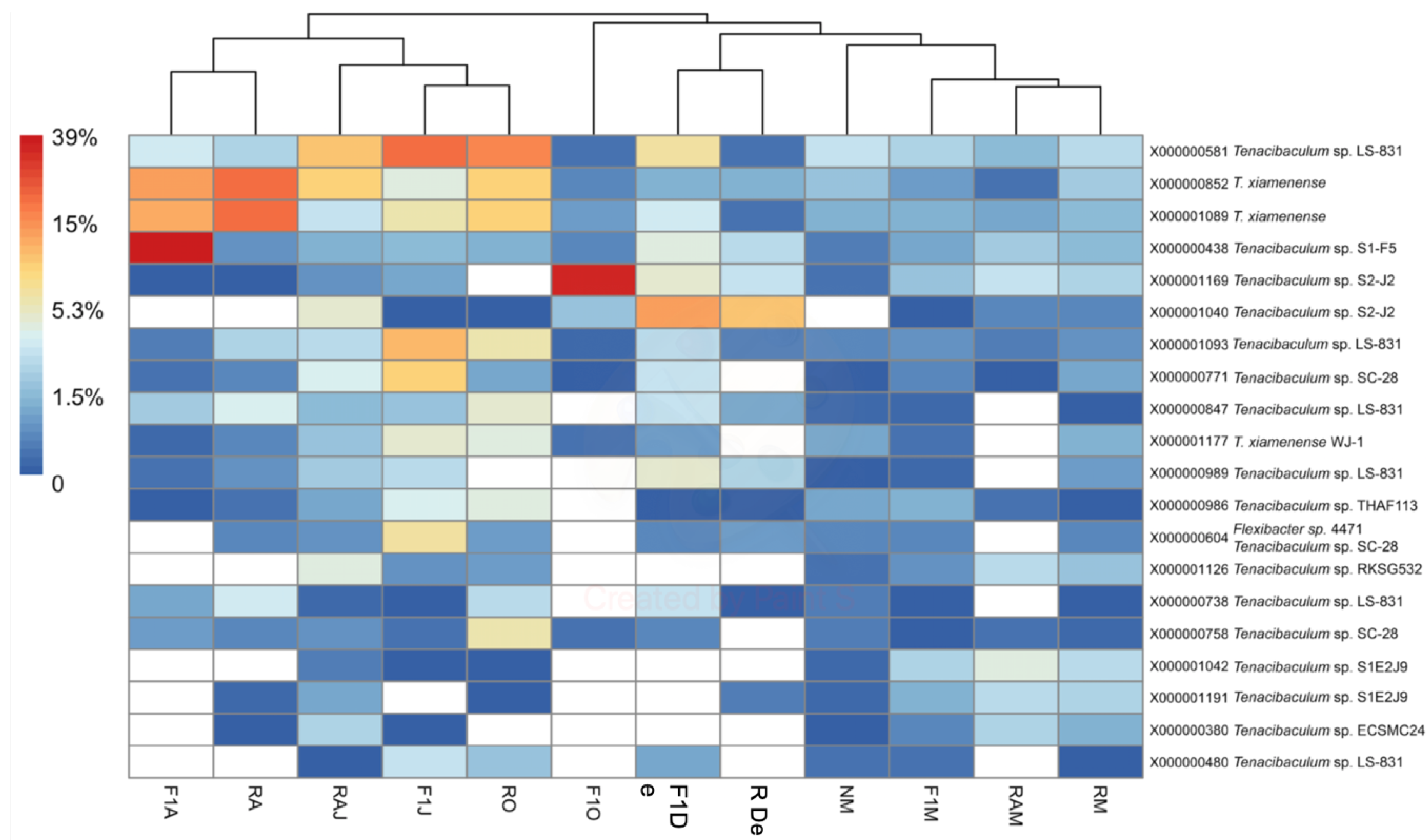
3.2. High-Throughput Sequence Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vega, R.; Pradenas, M.; Estrada, J.M.; Ramírez, D.; Valdebenito, I.; Mardones, A.; Dantagnan, P.; Alfaro, D.; Encina, F.; Pichara, C. Evaluación y comparación de la eficiencia de dos sistemas de incubación de huevos de Genypterus chilensis (Guichenot, 1848). LAJAR 2012, 40, 187–200. [Google Scholar] [CrossRef]
- Jara-Seguel, P.; Ubilla, A.; Estrada, J.; Ramírez, D.; Valdebenito, I. Nuclear DNA content in the red conger eel Genypterus chilensis (Guichenot, 1881) (Actinopterygii: Ophidiidae). Gayana 2011, 75, 198–200. [Google Scholar] [CrossRef]
- Levican, A.; Avendano-Herrera, R. Bacteria associated with mass mortality of post-larvae of red conger eel (Genypterus chilensis) cultured in a Chilean farm. Bull. Eur. Assoc. Fish Pathol. 2015, 35, 162–169. [Google Scholar]
- Lasa, A.; Avendaño-Herrera, R.; Estrada, J.M.; Romalde, J.L. Isolation and identification of Vibrio toranzoniae associated with diseased red conger eel (Genypterus chilensis) farmed in Chile. Vet. Microbiol. 2015, 179, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Levican, A.; Lasa, A.; Irgang, R.; Romalde, J.L.; Poblete-Morales, M.; Avendaño-Herrera, R. Isolation of Vibrio tapetis from two native fish species (Genypterus chilensis and Paralichthys adspersus) reared in Chile and description of Vibrio tapetis subsp. quintayensis subsp. nov. Int. J. Syst. Evol. Microbiol. 2017, 67, 716–723. [Google Scholar] [CrossRef]
- Irgang, R.; González-Luna, R.; Gutiérrez, J.; Poblete-Morales, M.; Rojas, V.; Tapia-Cammas, D.; Avendaño-Herrera, R. First identification and characterization of Tenacibaculum dicentrarchi isolated from Chilean red conger eel (Genypterus chilensis, Guichenot 1848). J. Fish Dis. 2017, 40, 1915–1920. [Google Scholar] [CrossRef]
- Giatsis, C.; Sipkema, D.; Smidt, H.; Verreth, J.; Verdegem, M. The colonization dynamics of the gut microbiota in tilapia larvae. PLoS ONE 2014, 9, e103641. [Google Scholar] [CrossRef]
- Taniguchi, A.; Aoki, R.; Eguchi, M. Microbial communities in various waters used for fish larval rearing. Aquac. Res. 2016, 47, 370–378. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. Fine Flounder (Paralichthys adspersus) Microbiome showed important differences between wild and reared specimens. Front. Microbiol. 2017, 8, 271. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. The microbiome of Seriola lalandi of wild and aquaculture origin reveals differences in composition and potential function. Front. Microbiol. 2017, 8, 1844. [Google Scholar] [CrossRef]
- DGAC. Dirección General de Aeronáutica Civil, Dirección Meteorológica de Chile, Subdepartamento climatología y meteorología aplicada, 2015. In Anuario Climatológico 2014; Estación Central: Santiago, Chile, 2015; ISSN 0716.3274. [Google Scholar]
- DGAC. Dirección General de Aeronáutica Civil, Dirección Meteorológica de Chile, Subdepartamento climatología y meteorología aplicada, 2016. In Anuario Climatológico 2015; Estación Central: Santiago, Chile, 2016; ISSN 0716.3274. [Google Scholar]
- Versalovic, J.; Koeuth, T.; Lupski, J.R. Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nuc. Acid Res. 1991, 19, 6283–6831. [Google Scholar] [CrossRef]
- Edwards, U.; Rogall, T.; Bloecker, H.; Emde, M.; Boettger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Lee, J.H.; Jung, Y.; Kim, M.; Kim, S.; Kim, B.K.; Lim, Y.W. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2259–2261. [Google Scholar] [CrossRef] [PubMed]
- MacFaddin, J.F. Pruebas Bioquímicas para la Identificación de Bacterias de Importancia Clínica; Editorial Médica Panamericana: Buenos Aires, Argentina, 2003. [Google Scholar]
- CLSI, Clinical and Laboratory Standard Institute. Performance Standards for Antimicrobial Susceptibility Testing of Bacteria Isolated from Aquatic Animals; Second Informational Supplement; VET03/VET04-S2; Clinical and Laboratory Standard Institute: Wayne, PA, USA, 2014. [Google Scholar]
- Huse, S.M.; Young, V.B.; Morrison, H.G.; Antonopoulos, D.A.; Kwon, J.; Dalal, S.; Arrieta, R.; Hubert, N.A.; Shen, L.; Vineis, J.H.; et al. Comparison of brush and biopsy sampling methods of the ileal pouch for assessment of mucosa-associated microbiota of human subjects. Microbiome 2014, 2, 5. [Google Scholar] [CrossRef]
- Eren, A.M.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L. A filtering method to generate high quality short reads using Illumina paired-end technology. PLoS ONE 2013, 8, e66643. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Donovan, H.P.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Eren, A.M.; Morrison, H.G.; Lescault, P.J.; Reveillaud, J.; Vineis, J.H.; Sogin, M.L. Minimum entropy decomposition: Unsupervised oligotyping for sensitive partitioning of high-throughput marker gene sequences. ISME J. 2015, 9, 968–979. [Google Scholar] [CrossRef]
- Marcon, E.; Herault, B. entropart: An R Package to Measure and Partition Diversity. J. Stat. Softw. 2015, 67, 1–26. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org/ (accessed on 16 June 2020).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-1. 2016. Available online: https://CRAN.R-project.org/package=vegan (accessed on 16 June 2020).
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.8. 2015. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 16 June 2020).
- Avendaño-Herrera, R.; Toranzo, A.E.; Magariños, B. Tenacibaculosis infection in marine fish caused by Tenacibaculum maritimum: A review. Dis. Aquat. Organ. 2006, 71, 255–266. [Google Scholar] [CrossRef]
- Hiergeist, A.; Gläsner, J.; Reischl, U.; Gessner, A. Analyses of intestinal microbiota: Culture versus sequencing. ILAR J. 2015, 56, 228–240. [Google Scholar] [CrossRef]
- Moreno, M.L.; Landgraf, M. Virulence factors and pathogenicity of Vibrio vulnificus strains isolated from seafood. J. Appl. Microbiol. 1998, 84, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Huang, L.; Su, Y.; Qin, Y.; Zhao, L.; Yan, Q. secA, secD, secF, yajC and yidC contribute to the adhesion regulation of Vibrio alginolyticus. Microbiologyopen 2018, 7, e551. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Akagawa-Matsushita, M.; Muroga, K. Vibrio ichthyoenteri sp. nov., a pathogen of Japanese flounder (Paralichthys olivaceus) larvae. Int. J. Syst. Bacteriol. 1996, 46, 155–159. [Google Scholar] [CrossRef]
- Kim, M.S.; Cho, J.Y.; Choi, H.S. Identification of Vibrio harveyi, Vibrio ichthyoenteri, and Photobacterium damselae isolated from olive flounder Paralichthys olivaceus in Korea by multiplex PCR developed using the rpoB gene. Fish. Sci. 2014, 80, 333–339. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; Leadbeater, S.; Garcia, C.; Sylvain, F.E.; Custodio, M.; Ang, K.P.; Powell, F.; Carvalho, G.R.; Creer, S.; Elliot, J.; et al. Parasitism perturbs the mucosal microbiome of Atlantic Salmon. Sci. Rep. 2017, 7, 43465. [Google Scholar] [CrossRef]
- Hooper, R.; Brealey, J.C.; van der Valk, T.; Alberdi, A.; Durban, J.W.; Fearnbach, H.; Robertson, K.M.; Baird, R.W.; Hanson, B.; Wade, P.; et al. Host-derived population genomics data provides insights into bacterial and diatom composition of the killer whale skin. Mol. Ecol. 2019, 28, 484–502. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Temp Max | ID Number | Genus | Species | Isolation Media | LIP | GEL | AMY |
|---|---|---|---|---|---|---|---|---|
| F1 Mar | 21.7 °C March 2014 | Q24 | Pseudoalteromonas | P. carragenovora | TSA-1 | - | + | - |
| Q25 | Pseudoalteromonas | P. undina | TSA-1 | - | + | - | ||
| Q27 | Psychrobacter | P. piscatori | TCBS | - | - | - | ||
| RA Mar | 21.7 °C March 2014 | Q1 | Pseudoalteromonas | P. espejiana | TSA-1 | - | + | - |
| Q2 | Pseudoalteromonas | P. espejiana | TSA-1 | - | - | - | ||
| Q3 | Aliivibrio | A. logei | TCBS | - | - | - | ||
| R Mar | 21.7 °C March 2014 | Q12 | Psychromonas | P. ingahanii | TSA-1 | - | - | - |
| Q14 | Shewanella | S. pacifica | TSA-1 | - | + | - | ||
| Q15 | Photobacterium | P. aquimaris | TSA-1 | - | - | - | ||
| Q16 | Vibrio | V. cortegadensis | TCBS | - | - | - | ||
| N Mar | 21.7 °C March 2014 | Q38 | Vibrio | V. hemicentroti | TSA-1 | - | + | - |
| Q39 | Idiomarina | I. loihensis | TSA-1 | - | + | - | ||
| Q40 | Alteromonas | Alteromonas sp. | TSA-1 | - | - | - | ||
| F1 June | 14.8 °C June 2014 | Q64 | Vibrio | V. lentus | TSA-1 | + | - | - |
| Q65 | Alivibrio | A. sifiae | TSA-1 | + | - | - | ||
| Q66 | Shewanella | S. pneumatophori | TSA-1 | + | - | - | ||
| Q67 | Alivibrio | A. sifiae | TCBS | - | - | - | ||
| Q68 | Paraperlucidibaca | P. baekdonensis | TSA-1 | - | - | - | ||
| Q69 | Alteromonas | A. australica | TSA-1 | + | - | - | ||
| RA June | 14.8 °C June 2014 | Q59 | Pseudoalteromonas | P. arctica | TSA-1 | + | - | - |
| Q60 | Shewanella | S. sairae | TSA-1 | + | - | - | ||
| Q61 | Psychromonas | P. arctica | TSA-1 | - | - | - | ||
| Q62 | Alivibrio | A. sifiae | TCBS | + | - | - | ||
| F1 Oct | 18.9 °C October 2014 | Q77 | Vibrio | V. gallaecicus | TSA-1 | - | - | - |
| RA Oct | 18.9 °C# October 2014 | Q70 | Vibrio | V. hemicentroti | TSA-1 | - | + | + |
| Q71 | Pseudoalteromonas | P. arctica | TSA-1 | + | - | - | ||
| Q72 | Shewanella | S. schegeliana | TSA-1 | - | + | - | ||
| Q73 | Bacillus | B. hwajinpoensis | TSA-1 | - | + | + | ||
| Q74 | Bacillus | B. thioparans | TSA-1 | - | - | + | ||
| Q75 | Paraperlucidibaca | P. baekdonensis | TSA-1 | - | - | - | ||
| R Oct | 18.9 °C October 2014 | Q76 | Bacillus | B. toyonensis | TSA-1 | - | + | - |
| F1 Dec | 21.2 °C December 2014 | Q101 | Vibrio | Vibrio sp. | TSA-1 | - | - | + |
| Q102 | Vibrio | V. lentus | TSA-1 | - | - | + | ||
| Q103 | Vibrio | V. splendidus | TSA-1 | - | - | + | ||
| Q104 | Vibrio | Vibrio sp. | TSA-1/TCBS | - | - | + | ||
| R Dec | 21.2 °C December 2014 | Q94 | Aliivibrio | A. finisterrensis | TSA-1 | - | - | - |
| Q95 | Photobacterium | P. piscicola | TSA-1 | + | - | - | ||
| Q96 | Alivibrio | A. sifiae | TSA-1 | + | - | - | ||
| Q97 | Vibrio | V. tapetis | TSA-1 | - | - | - | ||
| Q98 | Vibrio | V. gigantis | TCBS | - | - | - | ||
| F1 Apr | 21.3 °C April 2015 | Q156 | Psychromonas | P. aquatilis | TSA-1 | - | - | - |
| Q157 | Psychrobacter | P. proteolyticus | TSA-1 | - | - | - | ||
| Q158 | Photobacterium | P. sanguinicancri | TSA-1 | + | - | + | ||
| Q165 | Pseudoalteromonas | P. marina | TSA-1 | - | - | - | ||
| Q168 | Vibrio | V. splendidus | TCBS | - | - | + | ||
| Q160 | Acinetobacter | A. ursingii | TSA-1 | - | - | - | ||
| R Apr | 21.3 °C April 2015 | Q151 | Alivibrio | A. logei | TSA-1 | - | - | - |
| Q152 | Photobacterium | P. aquimaris | TSA-1 | - | - | - | ||
| Q153 | Acinetobacter | A. lwofii | TSA-1 | - | - | - | ||
| Q155 | Vibrio | V. splendidus | TCBS | - | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levican, A.; Fisher, J.C.; McLellan, S.L.; Avendaño-Herrera, R. Microbial Communities Associated with Farmed Genypterus chilensis: Detection in Water Prior to Bacterial Outbreaks Using Culturing and High-Throughput Sequencing. Animals 2020, 10, 1055. https://doi.org/10.3390/ani10061055
Levican A, Fisher JC, McLellan SL, Avendaño-Herrera R. Microbial Communities Associated with Farmed Genypterus chilensis: Detection in Water Prior to Bacterial Outbreaks Using Culturing and High-Throughput Sequencing. Animals. 2020; 10(6):1055. https://doi.org/10.3390/ani10061055
Chicago/Turabian StyleLevican, Arturo, Jenny C. Fisher, Sandra L. McLellan, and Ruben Avendaño-Herrera. 2020. "Microbial Communities Associated with Farmed Genypterus chilensis: Detection in Water Prior to Bacterial Outbreaks Using Culturing and High-Throughput Sequencing" Animals 10, no. 6: 1055. https://doi.org/10.3390/ani10061055
APA StyleLevican, A., Fisher, J. C., McLellan, S. L., & Avendaño-Herrera, R. (2020). Microbial Communities Associated with Farmed Genypterus chilensis: Detection in Water Prior to Bacterial Outbreaks Using Culturing and High-Throughput Sequencing. Animals, 10(6), 1055. https://doi.org/10.3390/ani10061055