Pharmacokinetic Profile of Oral Administration of Mefloquine to Clinically Normal Cats: A Preliminary In-Vivo Study of a Potential Treatment for Feline Infectious Peritonitis (FIP)


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drug Administration and Sample Collection
2.3. Drug Analysis Method and Sample Processing
2.3.1. Chemicals
2.3.2. HPLC Conditions
2.4. Pharmacokinetic Analysis
2.5. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Felten, S.; Hartmann, K. Diagnosis of Feline Infectious Peritonitis: A Review of the Current Literature. Viruses 2019, 11, 1068. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Allen, C.E.; Lyons, L.A. Pathogenesis of feline enteric coronavirus infection. J. Feline Med. Surg. 2008, 10, 529–541. [Google Scholar] [CrossRef]
- Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; Lutz, H.; et al. Feline infectious peritonitis. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 594–604. [Google Scholar] [CrossRef]
- Pedersen, N.C. A review of feline infectious peritonitis virus infection: 1963–2008. J. Feline Med. Surg. 2009, 11, 225–258. [Google Scholar] [CrossRef]
- Pedersen, N.C. An update on feline infectious peritonitis: Diagnostics and therapeutics. Vet. J. 2014, 201, 133–141. [Google Scholar] [CrossRef]
- Tsai, H.Y.; Chueh, L.L.; Lin, C.N.; Su, B.L. Clinicopathological findings and disease staging of feline infectious peritonitis: 51 cases from 2003 to 2009 in Taiwan. J. Feline Med. Surg. 2011, 13, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Hugo, T.B.; Heading, K.L. Prolonged survival of a cat diagnosed with feline infectious peritonitis by immunohistochemistry. Can. Vet. J. 2015, 56, 53–58. [Google Scholar] [PubMed]
- Ishida, T.; Shibanai, A.; Tanaka, S.; Uchida, K.; Mochizuki, M. Use of recombinant feline interferon and glucocorticoid in the treatment of feline infectious peritonitis. J. Feline Med. Surg. 2004, 6, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog. 2016, 12, e1005531. [Google Scholar]
- Kim, Y.; Mandadapu, S.R.; Groutas, W.C.; Chang, K.O. Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antiviral Res. 2013, 97, 161–168. [Google Scholar] [CrossRef]
- Murphy, B.G.; Perron, M.; Murakami, E.; Bauer, K.; Park, Y.; Eckstrand, C.; Liepnieks, M.; Pedersen, N.C. The nucleoside analog GS-441524 strongly inhibits feline infectious peritonitis (FIP) virus in tissue culture and experimental cat infection studies. Vet. Microbiol. 2018, 219, 226–233. [Google Scholar] [CrossRef]
- Pedersen, N.C.; Kim, Y.; Liu, H.; Galasiti Kankanamalage, A.C.; Eckstrand, C.; Groutas, W.C.; Bannasch, M.; Meadows, J.M.; Chang, K.O. Efficacy of a 3C-like protease inhibitor in treating various forms of acquired feline infectious peritonitis. J. Feline Med. Surg. 2018, 20, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Perron, M.; Bannasch, M.; Montgomery, E.; Murakami, E.; Liepnieks, M.; Liu, H. Efficacy and safety of the nucleoside analog GS-441524 for treatment of cats with naturally occurring feline infectious peritonitis. J. Feline Med. Surg. 2019, 21, 271–281. [Google Scholar] [CrossRef]
- Hsieh, L.E.; Lin, C.N.; Su, B.L.; Jan, T.R.; Chen, C.M.; Wang, C.H.; Lin, D.S.; Lin, C.T.; Chueh, L.L. Synergistic antiviral effect of Galanthus nivalis agglutinin and nelfinavir against feline coronavirus. Antiviral Res. 2010, 88, 25–30. [Google Scholar] [CrossRef]
- Strasfeld, L.; Chou, S. Antiviral drug resistance: Mechanisms and clinical implications. Infect. Dis. Clin. North. Am. 2010, 24, 413–437. [Google Scholar] [CrossRef] [PubMed]
- Radford, A.D.; Addie, D.; Belak, S.; Boucraut-Baralon, C.; Egberink, H.; Frymus, T.; Gruffydd-Jones, T.; Hartmann, K.; Hosie, M.J.; Lloret, A.; et al. ABCD guidelines on prevention and management. J. Feline Med. Surg. 2009, 11, 556–564. [Google Scholar] [CrossRef]
- McDonagh, P.; Sheehy, P.A.; Norris, J.M. Identification and characterisation of small molecule inhibitors of feline coronavirus replication. Vet. Microbiol. 2014, 174, 438–447. [Google Scholar] [CrossRef]
- Izes, A.M.; Kimble, B.; Norris, J.M.; Govendir, M. In vitro hepatic metabolism of mefloquine using microsomes from cats, dogs and the common brush-tailed possum (Trichosurus vulpecula). PLoS ONE 2020, 15, e0230975. [Google Scholar] [CrossRef]
- Izes, A.M. Comparative Studies of in Vitro Hepatic Metabolism of Mefloquine by Feline Microsomes and Those of Other Selected Species. Ph.D. Thesis, The University of Sydney, Camperdown, NSW, Australia, November 2019. [Google Scholar]
- Schwartz, D.E.; Weber, W.; Richard-Lenoble, D.; Gentilini, M. Kinetic studies of mefloquine and of one of its metabolites, Ro 21-5104, in the dog and in man. Acta. Trop. 1980, 37, 238–242. [Google Scholar] [PubMed]
- Remple, J.D. Intracellular Hematozoa of Raptors: A Review and Update. J. Avian Med. Surg. 2004, 18, 75–88. [Google Scholar] [CrossRef]
- Grilo, M.L.; Vanstreels, R.E.; Wallace, R.; Garcia-Parraga, D.; Braga, E.M.; Chitty, J.; Catao-Dias, J.L.; Madeira de Carvalho, L.M. Malaria in penguins—Current perceptions. Avian Pathol. 2016, 45, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Suzuki, K.; Fujita, H.; Uzuka, T.; Matsuda, H.; Shishido-Hara, Y.; Arai, S.; Nakamura, T.; Kikuchi, S.; Nakamichi, K.; et al. Successful treatment of non-HIV progressive multifocal leukoencephalopathy: Case report and literature review. J. Neurol. 2020, 267, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Nambirajan, A.; Suri, V.; Kataria, V.; Sharma, M.C.; Goyal, V. Progressive multifocal leukoencephalopathy in a 44-year old male with idiopathic CD4+ T-lymphocytopenia treated with mirtazapine and mefloquine. Neurol. India 2017, 65, 1061–1064. [Google Scholar] [PubMed]
- McDonagh, P.; Sheehy, P.A.; Fawcett, A.; Norris, J.M. Antiviral effect of mefloquine on feline calicivirus in vitro. Vet. Microbiol. 2015, 176, 370–377. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Teramoto, T.; Kulkarni, A.A.; Bhattacharjee, A.K.; Padmanabhan, R. Antiviral activities of selected antimalarials against dengue virus type 2 and Zika virus. Antiviral Res. 2017, 137, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.H.; Wang, L.Q.; Liu, W.L.; An, X.P.; Liu, Z.D.; He, X.Q.; Song, L.H.; Tong, Y.G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus (2019-nCoV) related coronavirus model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef]
- Brickelmaier, M.; Lugovskoy, A.; Kartikeyan, R.; Reviriego-Mendoza, M.M.; Allaire, N.; Simon, K.; Frisque, R.J.; Gorelik, L. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob. Agents Chemother. 2009, 53, 1840–1849. [Google Scholar] [CrossRef]
- Owen, A.; Janneh, O.; Hartkoorn, R.C.; Chandler, B.; Bray, P.G.; Martin, P.; Ward, S.A.; Hart, C.A.; Khoo, S.H.; Back, D.J. In vitro synergy and enhanced murine brain penetration of saquinavir coadministered with mefloquine. J. Pharmacol. Exp. Ther. 2005, 314, 1202–1209. [Google Scholar] [CrossRef]
- Palmer, K.J.; Holliday, S.M.; Brogden, R.N. Mefloquine. A review of its antimalarial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1993, 45, 430–475. [Google Scholar] [CrossRef]
- Karbwang, J.; White, N.J. Clinical pharmacokinetics of mefloquine. Clin. Pharmacokinet. 1990, 19, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, R.E.; Pamplin, C.L., III; von Bredow, J.; Barry, K.G.; Canfield, C.J. Kinetics of a new antimalarial, mefloquine. Clin. Pharmacol. Ther. 1979, 26, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Karbwang, J.; Na-Bangchang, K. Clinical application of mefloquine pharmacokinetics in the treatment of P falciparum malaria. Fundam. Clin. Pharmacol. 1994, 8, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Juma, F.D.; Ogeto, J.O. Mefloquine disposition in normals and in patients with severe Plasmodium falciparum malaria. Eur. J. Drug Metab. Pharmacokinet. 1989, 14, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.R.; Arguin, P.M.; Arguin, T. Travel Related Infectious Disease. In Centers for Disease Control and PRevention. CDC Yellow Book 2020: Health Information for International Travel. Available online: https://www.cbc.gov/malaria/travelers/drugs.html (accessed on 21 April 2020).
- Centers for Disease Control and Prevention, Malaria, how to Choose a Drug to Prevent Malaria. Available online: https://www.cdc.gov/malaria/travelers/drugs.html (accessed on 21 April 2020).
- White, N.J. Clinical pharmacokinetics of antimalarial drugs. Clin. Pharmacokinet. 1985, 10, 187–215. [Google Scholar] [CrossRef]
- Crevoisier, C.; Handschin, J.; Barre, J.; Roumenov, D.; Kleinbloesem, C. Food increases the bioavailability of mefloquine. Eur. J. Clin. Pharmacol. 1997, 53, 135–139. [Google Scholar] [CrossRef]
- Antony, H.A.; Parija, S.C. Antimalarial drug resistance: An overview. Trop. Parasitol. 2016, 6, 30–41. [Google Scholar]
- Takano, T.; Katoh, Y.; Doki, T.; Hohdatsu, T. Effect of chloroquine on feline infectious peritonitis virus infection in vitro and in vivo. Antiviral Res. 2013, 99, 100–107. [Google Scholar] [CrossRef]
- Lundstrom, K. Coronavirus Pandemic-Therapy and Vaccines. Biomedicines 2020, 8, 109. [Google Scholar] [CrossRef]
- Takano, T.; Satoh, K.; Doki, T.; Tanabe, T.; Hohdatsu, T. Antiviral Effects of Hydroxychloroquine and Type I Interferon on In Vitro Fatal Feline Coronavirus Infection. Viruses 2020, 12, 576. [Google Scholar] [CrossRef]
- Lobel, H.O.; Bernard, K.W.; Williams, S.L.; Hightower, A.W.; Patchen, L.C.; Campbell, C.C. Effectiveness and tolerance of long-term malaria prophylaxis with mefloquine. Need for a better dosing regimen. JAMA 1991, 265, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Tin, F.; Hlaing, N.; Lasserre, R. Single-dose treatment of falciparum malaria with mefloquine: Field studies with different doses in semi-immune adults and children in Burma. Bull. World Health Organ. 1982, 60, 913–917. [Google Scholar] [PubMed]
- Lobel, H.O.; Miani, M.; Eng, T.; Bernard, K.W.; Hightower, A.W.; Campbell, C.C. Long-term malaria prophylaxis with weekly mefloquine. Lancet 1993, 341, 848–851. [Google Scholar] [CrossRef]
- Gribble, F.M.; Davis, T.M.; Higham, C.E.; Clark, A.; Ashcroft, F.M. The antimalarial agent mefloquine inhibits ATP-sensitive K-channels. Br. J. Pharmacol. 2000, 131, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Ringqvist, A.; Bech, P.; Glenthoj, B.; Petersen, E. Acute and long-term psychiatric side effects of mefloquine: A follow-up on Danish adverse event reports. Travel Med. Infect. Dis. 2015, 13, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, E.C.; Block, J.; Nevin, R.L. Psychiatric side effects of mefloquine: Applications to forensic psychiatry. J. Am. Acad. Psychiatry Law 2013, 41, 224–235. [Google Scholar]
- Lee, S.J.; Ter Kuile, F.O.; Price, R.N.; Luxemburger, C.; Nosten, F. Adverse effects of mefloquine for the treatment of uncomplicated malaria in Thailand: A pooled analysis of 19, 850 individual patients. PLoS ONE 2017, 12, e0168780. [Google Scholar] [CrossRef]
- Smith, H.R.; Croft, A.M.; Black, M.M. Dermatological adverse effects with the antimalarial drug mefloquine: A review of 74 published case reports. Clin. Exp. Dermatol. 1999, 24, 249–254. [Google Scholar] [CrossRef]
- Stracher, A.R.; Stoeckle, M.Y.; Giordano, M.F. Aplastic anemia during malarial prophylaxis with mefloquine. Clin. Infect. Dis. 1994, 18, 263–264. [Google Scholar] [CrossRef]
- Magnussen, P.; Bygbjerg, I.C. Treatment of Plasmodium falciparum malaria with mefloquine alone or in combination with i.v. quinine at the Department of Communicable and Tropical Diseases, Rigshospitalet, Copenhagen 1982–1988. Dan. Med. Bull. 1990, 37, 563–564. [Google Scholar]
- Klotz, U. Pharmacokinetics and drug metabolism in the elderly. Drug Metab. Rev. 2009, 41, 67–76. [Google Scholar] [CrossRef]
- Mangoni, A.A.; Jackson, S.H. Age-related changes in pharmacokinetics and pharmacodynamics: Basic principles and practical applications. Br. J. Clin. Pharmacol. 2004, 57, 6–14. [Google Scholar] [CrossRef]
- Singhasivanon, V.; Chongsuphajaisiddhi, T.; Sabcharoen, A.; Attanath, P.; Webster, H.K.; Wernsdorfer, W.H.; Sheth, U.K.; Djaja Lika, I. Pharmacokinetics of mefloquine in children aged 6 to 24 months. Eur. J. Drug Metab. Pharmacokinet. 1992, 17, 275–279. [Google Scholar] [CrossRef]
- Vieira, J.L.; Borges, L.M.; Ferreira, M.V.; Rivera, J.G.; Gomes Mdo, S. Patient age does not affect mefloquine concentrations in erythrocytes and plasma during the acute phase of falciparum malaria. Braz. J. Infect. Dis. 2016, 20, 482–486. [Google Scholar] [CrossRef]
- Karbwang, J.; Bunnag, D.; Breckenridge, A.M.; Back, D.J. The pharmacokinetics of mefloquine when given alone or in combination with sulphadoxine and pyrimethamine in Thai male and female subjects. Eur. J. Clin. Pharmacol. 1987, 32, 173–177. [Google Scholar] [CrossRef]
- Looareesuwan, S.; White, N.J.; Warrell, D.A.; Forgo, I.; Dubach, U.G.; Ranalder, U.B.; Schwartz, D.E. Studies of mefloquine bioavailability and kinetics using a stable isotope technique: A comparison of Thai patients with falciparum malaria and healthy Caucasian volunteers. Br. J. Clin. Pharmacol. 1987, 24, 37–42. [Google Scholar] [CrossRef]
- Fontaine, F.; de Sousa, G.; Burcham, P.C.; Duchene, P.; Rahmani, R. Role of cytochrome P450 3A in the metabolism of mefloquine in human and animal hepatocytes. Life Sci. 2000, 66, 2193–2212. [Google Scholar] [CrossRef]
- Shah, S.S.; Sanda, S.; Regmi, N.L.; Sasaki, K.; Shimoda, M. Characterization of cytochrome P450-mediated drug metabolism in cats. J. Vet. Pharmacol. Ther. 2007, 30, 422–428. [Google Scholar] [CrossRef]
- Reuter, S.E.; Upton, R.N.; Evans, A.M.; Navaratnam, V.; Olliaro, P.L. Population pharmacokinetics of orally administered mefloquine in healthy volunteers and patients with uncomplicated Plasmodium falciparum malaria. J. Antimicrob. Chemother. 2015, 70, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Zsila, F.; Visy, J.; Mady, G.; Fitos, I. Selective plasma protein binding of antimalarial drugs to alpha1-acid glycoprotein. Bioorg. Med. Chem. 2008, 16, 3759–3772. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, M.E.; Whicher, J.T.; Harbour, D.A. Cats inoculated with feline infectious peritonitis virus exhibit a biphasic acute phase plasma protein response. Vet. Rec. 1988, 123, 622–624. [Google Scholar] [PubMed]
- Duthie, S.; Eckersall, P.D.; Addie, D.D.; Lawrence, C.E.; Jarrett, O. Value of alpha 1-acid glycoprotein in the diagnosis of feline infectious peritonitis. Vet. Rec. 1997, 141, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Spagnolo, V.; Colombo, A.; Paltrinieri, S. Changes in some acute phase protein and immunoglobulin concentrations in cats affected by feline infectious peritonitis or exposed to feline coronavirus infection. Vet. J. 2004, 167, 38–44. [Google Scholar] [CrossRef]
- Paltrinieri, S.; Giordano, A.; Tranquillo, V.; Guazzetti, S. Critical assessment of the diagnostic value of feline alpha1-acid glycoprotein for feline infectious peritonitis using the likelihood ratios approach. J. Vet. Diagn. Investig. 2007, 19, 266–272. [Google Scholar]
- Hall, J.A.; Yerramilli, M.; Obare, E.; Yerramilli, M.; Jewell, D.E. Comparison of serum concentrations of symmetric dimethylarginine and creatinine as kidney function biomarkers in cats with chronic kidney disease. J. Vet. Intern. Med. 2014, 28, 1676–1683. [Google Scholar] [CrossRef]
- Nabity, M.B.; Lees, G.E.; Boggess, M.M.; Yerramilli, M.; Obare, E.; Yerramilli, M.; Rakitin, A.; Aguiar, J.; Relford, R. Symmetric dimethylarginine assay validation, stability, and evaluation as a marker for the early detection of chronic kidney disease in dogs. J. Vet. Intern. Med. 2015, 29, 1036–1044. [Google Scholar] [CrossRef]
- Yerramilli, M.; Farace, G.; Quinn, J.; Yerramilli, M. Kidney disease and the nexus of chronic kidney disease and acute kidney injury: The role of novel biomarkers as early and accurate diagnostics. Vet. Clin. North. Am. Small Anim. Pract. 2016, 46, 961–993. [Google Scholar] [CrossRef]
- Dahlem, D.P.; Neiger, R.; Schweighauser, A.; Francey, T.; Yerramilli, M.; Obare, E.; Steinbach, S.M.L. Plasma symmetric dimethylarginine concentration in dogs with acute kidney injury and chronic kidney disease. J. Vet. Intern. Med. 2017, 31, 799–804. [Google Scholar] [CrossRef]
- Braff, J.; Obare, E.; Yerramilli, M.; Elliott, J.; Yerramilli, M. Relationship between serum symmetric dimethylarginine concentration and glomerular filtration rate in cats. J. Vet. Intern. Med. 2014, 28, 1699–1701. [Google Scholar] [CrossRef]
- Wiwanitkit, V. Antimalarial drug and renal toxicity. J. Nephropharmacol. 2016, 5, 11–12. [Google Scholar]
- Namba, S.; Kitamura, R.; Amaha, T.; Befu, M.; Zama, T.; Moriwaki, T.; Kumono, S.; Shichijo, S. Impact of general anesthesia on serum symmetric dimethylarginine concentration in cats. In Proceedings of the American Association of Feline Practitioners Conference 2018, Charlotte, NC, USA, 27–30 September 2018. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Mefloquine Plasma Concentrations ug/mL | |||||||
|---|---|---|---|---|---|---|---|
| H | Cat D | Cat A | Cat B | Cat G | Cat E | Cat C | Cat F |
| Male Neutered | Male Neutered | Female Neutered | Female Neutered | Male Neutered | Female Neutered | Female Neutered | |
| 0 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| 1 | 0.54 | 1.58 | 0.31 | 1.97 | 0.22 | 0.86 | - |
| 2 | 0.95 | 1.95 | 0.89 | 2.79 | 0.40 | 2.19 | - |
| 4 | 1.13 | 2.10 | 1.84 | 3.23 | 0.49 | - | - |
| 8 | 1.59 | 2.23 | 2.20 | 2.89 | 0.64 | - | - |
| 12 | 1.77 | 2.97 | 2.14 | 3.22 | 0.72 | - | - |
| 24 | 2.09 | 3.35 | 2.09 | 3.12 | 0.56 | - | - |
| 48 | 1.86 | 3.34 | 1.97 | 2.88 | 0.56 | - | - |
| 96 | 1.51 | 2.76 | 1.75 | 2.50 | 0.57 | - | - |
| 168 | 2.75 | 4.15 | 3.36 | 3.73 | 3.58 | - | 1.39 |
| 240 | 1.85 | 6.51 | 2.93 | 4.94 | 4.20 | - | 1.54 |
| 336 | 3.07 | 4.19 | 3.60 | 4.56 | 2.09 | - | 2.11 |
| PK Indices | Units | Mean | SD | Median | Min | Max |
|---|---|---|---|---|---|---|
| ke (48–96 h) | 1/h | 0.003 | 0.001 | 0.003 | 0.003 | 0.005 |
| t1/2 | h | 224.18 | 51.60 | 233.94 | 153.24 | 275.60 |
| Tmax | h | 15.00 | 10.52 | 16.00 | 4.00 | 24.00 |
| Cmax | μg/mL | 2.71 | 0.66 | 2.71 | 2.09 | 3.35 |
| AUC 0–96 h | μg/mL × h | 228.30 | 62.23 | 228.18 | 166.59 | 290.25 |
| AUMC 0–96 h | μg/mL × h2 | 10737 | 2971.7 | 10576 | 7826.0 | 13968 |
| MRT 0–96 h | h | 326.50 | 13.60 | 337.46 | 221.17 | 397.47 |
| V/Fobs (as calculated over 0–96 h) | L/kg | 17.41 | 4.08 | 15.74 | 14.73 | 23.41 |
| Cl/Fobs (as calculated over 0–96 h) | L/h/kg | 0.06 | 0.02 | 0.052 | 0.04 | 0.085 |
| Biochemical Analyte | 0 h | 168 h | 336 h | Reference Interval (Idexx Reference Laboratory) | ||||
|---|---|---|---|---|---|---|---|---|
| Units | Mean | Range | Mean | Range | Mean | Range | ||
| Glucose | mmol/L | - | - | 5.10 | 3.90–6.30 | 4.50 | 3.40–5.40 | 3.20–7.50 |
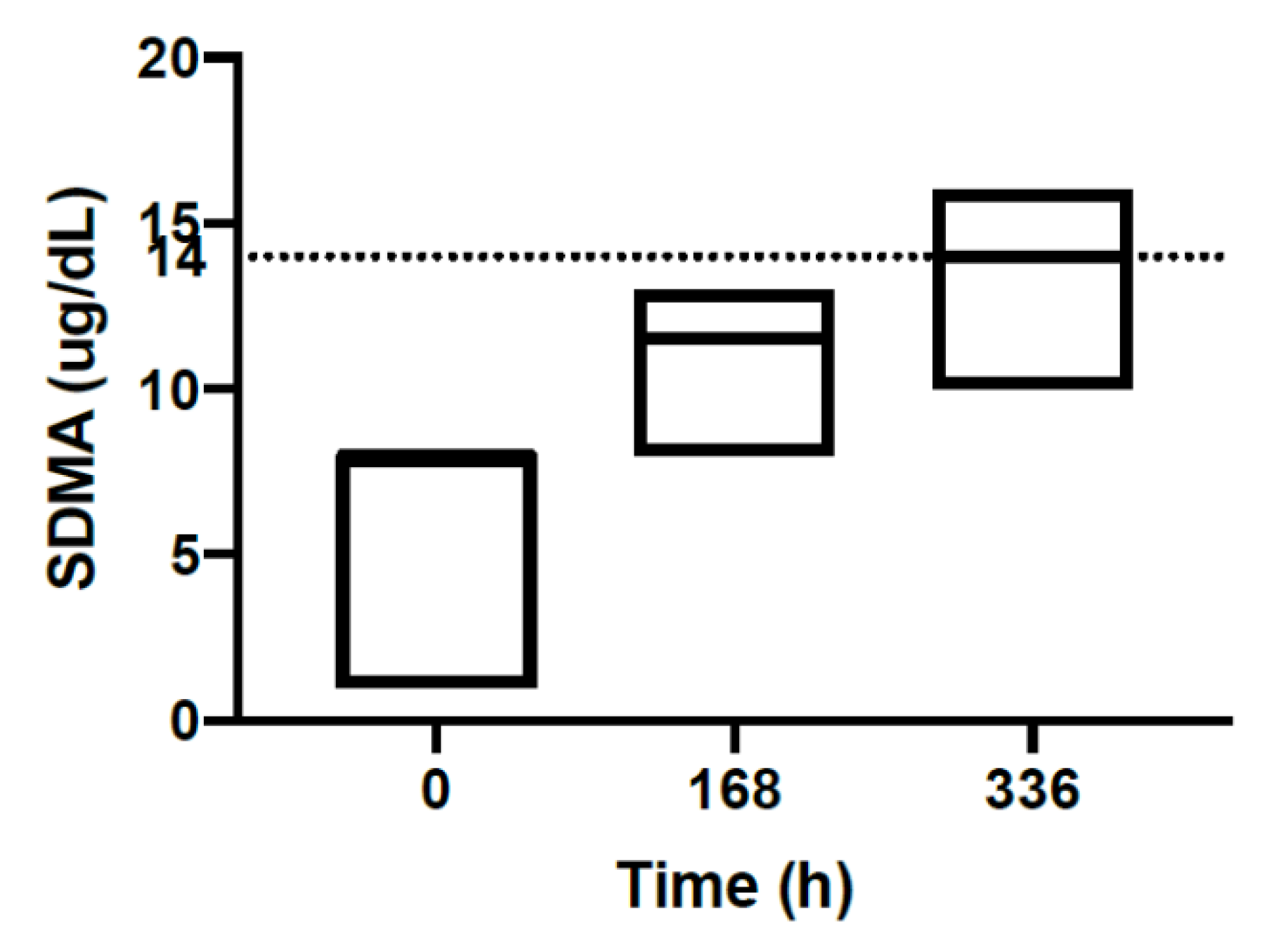
| SDMA | ug/dL | 6.70 | 1.00–8.00 | 11.0 | 8.00–13.0 | 13.5 | 10.0–16.0 | 0.00–14.0 |
| Creatinine | umol/L | 115 | 90.0–140. | 122 | 80.0–160. | 120. | 100.–140. | 80.0–200. |
| Urea | mmol/L | 8.00 | 6.80–10.2 | 7.70 | 6.90–9.10 | 8.08 | 6.90–9.20 | 5.00–15.0 |
| Phosphorus | mmol/L | 1.70 | 1.40–2.00 | 1.40 | 1.17–1.63 | 1.32 | 1.20–1.60 | 0.00–2.30 |
| Calcium | mmol/L | 2.40 | 2.30–2.50 | 2.40 | 2.40–2.60 | 2.30 | 2.20–2.40 | 2.10–2.80 |
| Sodium | mmol/L | 152 | 149–153 | 154 | 152–156 | 151 | 148–153 | 144–158 |
| Potassium | mmol/L | 5.10 | 4.50–5.20 | 4.50 | 4.10–5.20 | 4.40 | 4.10–4.70 | 3.70–5.40 |
| Chloride | mmol/L | 115 | 111–117 | 123 | 120–125 | 118 | 116–120 | 106–123 |
| Bicarbonate | mmol/L | 16.0 | 15.0–18.0 | - | - | 16.3 (4 cats) | 15.0–18.0 | 12.0–24.0 |
| Anion gap | mmol/L | 25.8 | 25.2–27.1 | - | - | 20.6 (4 cats) | 20.1–21.3 | 15.0–31.0 |
| Total protein | g/L | 75.3 | 67.0–86.0 | 71.0 | 65.2–80.7 | 71.3 | 65.0–83.7 | 60.0– 84.0 |
| Albumin | g/l | 31.7 | 29.0–36.0 | 28.9 | 27.8–30.0 | 30.0 | 28.0–32.0 | 25.0–38.0 |
| Globulin | g/L | 43.7 | 35.0–37.0 | 42.3 | 35.3–52.9 | 41.2 | 33.0–55.7 | 31.0–52.0 |
| ALT | U/L | 79.2 | 43.0–161 | 47.3 | 26.0–116 | 58.7 | 22.0–166 | 19.0–100. |
| AST | U/L | 47.8 | 25.0–83.0 | 29.7 | 22.0–53.0 | 29.8 | 20.0–42.0 | 0.00–62.0 |
| ALP | U/L | 39.8 | 21.0–75.0 | 37.0 | 24.0–72.0 | 41.8 | 27.0–87.0 | 5.00–50.0 |
| GGT | U/L | 0.70 | 0.00–1.00 | - | - | 0.50 | 0.00–2.00 | 0.00–5.00 |
| Total bilirubin | umol/L | 3.00 | 3.00 | 2.10 | 0.40–2.70 | 2.60 | 2.00–3.00 | 0.00–7.00 |
| Cholesterol | mmol/L | 3.85 | 2.60–4.90 | 3.90 | 3.00–4.80 | 3.10 | 0.00–4.70 | 2.20–5.50 |
| CK | U/L | 299 | 133–735 | 178 | 97.0–398 | 148 | 17.0–248 | 64.0–400 |
| TT4 | nmol/L | 32.5 | 21.0–39.0 | 32.6 | 28.6–36.7 | 32.4–35.4 | 10.0–60.0 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, J.; Kimble, B.; Norris, J.M.; Govendir, M. Pharmacokinetic Profile of Oral Administration of Mefloquine to Clinically Normal Cats: A Preliminary In-Vivo Study of a Potential Treatment for Feline Infectious Peritonitis (FIP). Animals 2020, 10, 1000. https://doi.org/10.3390/ani10061000
Yu J, Kimble B, Norris JM, Govendir M. Pharmacokinetic Profile of Oral Administration of Mefloquine to Clinically Normal Cats: A Preliminary In-Vivo Study of a Potential Treatment for Feline Infectious Peritonitis (FIP). Animals. 2020; 10(6):1000. https://doi.org/10.3390/ani10061000
Chicago/Turabian StyleYu, Jane, Benjamin Kimble, Jacqueline M. Norris, and Merran Govendir. 2020. "Pharmacokinetic Profile of Oral Administration of Mefloquine to Clinically Normal Cats: A Preliminary In-Vivo Study of a Potential Treatment for Feline Infectious Peritonitis (FIP)" Animals 10, no. 6: 1000. https://doi.org/10.3390/ani10061000
APA StyleYu, J., Kimble, B., Norris, J. M., & Govendir, M. (2020). Pharmacokinetic Profile of Oral Administration of Mefloquine to Clinically Normal Cats: A Preliminary In-Vivo Study of a Potential Treatment for Feline Infectious Peritonitis (FIP). Animals, 10(6), 1000. https://doi.org/10.3390/ani10061000