Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Sequence Data Generation
2.3. Identification of Single Nucleotide Variants (SNVs) between C. Nubiana and the Domestic Goat
2.4. Capra Nubiana and Capra Hircus Protein-Coding DNA Sequences (CDS)
2.5. Protein-Coding DNA Sequences (CDS) for Positive Selection Analysis
2.6. Single Gene Ortholog Identification
2.7. The dN/dS Analysis
2.8. Functional Annotation of the Positively Selected Genes (PSG) and Sites
3. Results
3.1. Genome Sequence and SNVs Calling
3.2. Positively Selected Genes in Capra Nubiana
3.3. Skin Development and Barrier Function Genes under Positive Selection in C. Nubiana
4. Discussion
4.1. Whole-Genome Mapping, Single Nucleotide Variant Calling, and Annotation
4.2. Positively Selected Genes in Capra Nubiana
4.3. Skin Development and Barrier Function Genes under Positive Selection in C. Nubiana
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
References
- Pidancier, N.; Jordan, S.; Luikart, G.; Taberlet, P. Evolutionary history of the genus Capra (Mammalia, Artiodactyla): Discordance between mitochondrial DNA and Y-chromosome phylogenies. Mol. Phylogenet. Evol. 2006, 40, 739–749. [Google Scholar] [CrossRef]
- Castelló, J.R.; Huffman, B.; Groves, C. Bovids of the World Antelopes, Gazelles, Cattle, Goats, Sheep, and Relatives; Princeton University Press: Princeton, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Shackleton, D.M.; Caprinae Specialist Group. Wild Sheep and Goats and their Relatives.Status Survey and Conservation Action Plan. for Caprinae; IUCN: Cambridge, UK, 1997. [Google Scholar]
- Alkon, P.U.; Harding, L.; Jdeidi, T.; Masseti, M.; Nader, I.; de Smet, K.; Cuzin, F.; Saltz, D. The IUCN Red List of Threatened Species. Capra Nubiana 2008, E.T3796A10084254. [Google Scholar] [CrossRef]
- Henry, B.K.; Eckard, R.J.; Beauchemin, K.A. Review: Adaptation of ruminant livestock production systems to climate changes. Animal 2018, 12, s445–s456. [Google Scholar] [CrossRef]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar]
- Yang, Z.; Nielsen, R. Codon-Substitution Models for Detecting Molecular Adaptation at Individual Sites Along Specific Lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Nielsen, R.; Yang, Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R.; Goldman, N.; Pedersen, A.-M.K. Codon-Substitution Models for Heterogeneous Selection Pressure at Amino Acid Sites. Genetics 2000, 155, 431–449. [Google Scholar] [PubMed]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar] [PubMed]
- Bedi, S.K.; Prasad, A.; Mathur, K.; Bhatnagar, S. Positive selection and evolution of dengue type-3 virus in the Indian subcontinent. J. Vector Borne Dis. 2013, 50, 188–196. [Google Scholar]
- Yang, Z. Maximum Likelihood Estimation on Large Phylogenies and Analysis of Adaptive Evolution in Human Influenza Virus A. J. Mol. Evol. 2000, 51, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C.; Smith, J.M.; Hurst, L.D.; Holden, M.T.G.; Cooper, J.E.; Smith, N.H.; Feil, E.J. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 2006, 239, 226–235. [Google Scholar] [PubMed]
- Soyer, Y.; Orsi, R.H.; Rodriguez-Rivera, L.D.; Sun, Q.; Wiedmann, M. Genome wide evolutionary analyses reveal serotype specific patterns of positive selection in selected Salmonella serotypes. BMC Evol. Biol. 2009, 9, 264. [Google Scholar] [CrossRef]
- Qian, J.; Liu, Y.; Chao, N.; Ma, C.; Chen, Q.; Sun, J.; Wu, Y. Positive selection and functional divergence of farnesyl pyrophosphate synthase genes in plants. BMC Mol. Biol. 2017, 18, 3. [Google Scholar] [CrossRef][Green Version]
- De La Torre, A.R.; Li, Z.; Van de Peer, Y.; Ingvarsson, P.K. Contrasting Rates of Molecular Evolution and Patterns of Selection among Gymnosperms and Flowering Plants. Mol. Biol. Evol. 2017, 34, 1363–1377. [Google Scholar] [CrossRef]
- You, H.; Liu, Y.; Minh, T.N.; Lu, H.; Zhang, P.; Li, W.; Xiao, J.; Ding, X.; Li, Q. Genome-wide identification and expression analyses of nitrate transporter family genes in wild soybean (Glycine soja). J. Appl. Genet. 2020. [Google Scholar] [CrossRef]
- Wu, H.; Guang, X.; Al-Fageeh, M.B.; Cao, J.; Pan, S.; Zhou, H.; Zhang, L.; Abutarboush, M.H.; Xing, Y.; Xie, Z.; et al. Camelid genomes reveal evolution and adaptation to desert environments. Nat. Commun. 2014, 5, 5188. [Google Scholar] [CrossRef]
- Han, M.V.; Demuth, J.P.; McGrath, C.L.; Casola, C.; Hahn, M.W. Adaptive evolution of young gene duplicates in mammals. Genome Res. 2009, 19, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.; Li, Z.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364. [Google Scholar] [CrossRef]
- Xu, S.; Tian, R.; Lin, Y.; Yu, Z.; Zhang, Z.; Niu, X.; Wang, X.; Yang, G. Widespread positive selection on cetacean TLR extracellular domain. Mol. Immunol. 2019, 106, 135–142. [Google Scholar] [CrossRef]
- McGowen, M.R.; Grossman, L.I.; Wildman, D.E. Dolphin genome provides evidence for adaptive evolution of nervous system genes and a molecular rate slowdown. Proc. Biol Sci. 2012, 279, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Roux, J.; Privman, E.; Moretti, S.; Daub, J.T.; Robinson-Rechavi, M.; Keller, L. Patterns of positive selection in seven ant genomes. Mol. Biol. Evol. 2014, 31, 1661–1685. [Google Scholar] [CrossRef]
- Tong, C.; Li, M. Transcriptomic signature of rapidly evolving immune genes in a highland fish. Fish Shellfish Immunol. 2020, 97, 587–592. [Google Scholar] [CrossRef]
- Freitas, L.; Mesquita, R.; Schrago, C. Survey for positively selected coding regions in the genome of the hematophagous tsetse fly Glossina morsitans identifies candidate genes associated with feeding habits and embryonic development. Genet. Mol. Biol. 2020, 43. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24. [Google Scholar] [CrossRef]
- Agaba, M.; Ishengoma, E.; Miller, W.C.; McGrath, B.C.; Hudson, C.N.; Bedoya Reina, O.C.; Ratan, A.; Burhans, R.; Chikhi, R.; Medvedev, P.; et al. Giraffe genome sequence reveals clues to its unique morphology and physiology. Nat. Commun. 2016, 7, 11519. [Google Scholar] [CrossRef]
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, L.; Chen, X.; Zhong, Y.; Yang, Y.; Xia, W.; Liu, C.; Zhu, W.; Wang, H.; Yan, B.; et al. Biological adaptations in the Arctic cervid, the reindeer (Rangifer tarandus). Science 2019, 364. [Google Scholar] [CrossRef]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 15 June 2018).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol 1990, 215. [Google Scholar] [CrossRef]
- Ward, N.; Moreno-Hagelsieb, G. Quickly Finding Orthologs as Reciprocal Best Hits with BLAT, LAST, and UBLAST: How Much Do We Miss? PLoS ONE 2014, 9, e101850. [Google Scholar] [CrossRef]
- Wernersson, R.; Pedersen, A.G. RevTrans: Multiple alignment of coding DNA from aligned amino acid sequences. Nucleic Acids Res. 2003, 31, 3537–3539. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Yang, Z.; dos Reis, M. Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 2011, 28, 1217–1228. [Google Scholar] [CrossRef]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 2002, 19, 950–958. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Shahi, P.; Huang, J.T.W.; Phan, N.N.; Sun, Z.; Lin, Y.-C.; Lai, M.-D.; Werb, Z. Systematic analysis of the achaete-scute complex-like gene signature in clinical cancer patients. Mol. Clin. Oncol. 2017, 6, 7–18. [Google Scholar] [CrossRef]
- Ball, D.W.; Azzoli, C.G.; Baylin, S.B.; Chi, D.; Dou, S.; Donis-Keller, H.; Cumaraswamy, A.; Borges, M.; Nelkin, B.D. Identification of a human achaete-scute homolog highly expressed in neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 1993, 90, 5648–5652. [Google Scholar] [CrossRef]
- Wu, L.; Yavas, G.; Hong, H.; Tong, W.; Xiao, W. Direct comparison of performance of single nucleotide variant calling in human genome with alignment-based and assembly-based approaches. Sci. Rep. 2017, 7, 10963. [Google Scholar] [CrossRef]
- Bertolini, F.; Scimone, C.; Geraci, C.; Schiavo, G.; Utzeri, V.; Chiofalo, V.; Fontanesi, L. Next Generation Semiconductor Based Sequencing of the Donkey (Equus asinus) Genome Provided Comparative Sequence Data against the Horse Genome and a Few Millions of Single Nucleotide Polymorphisms. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Mei, C.; Wang, H.; Zhu, W.; Wang, H.; Cheng, G.; Qu, K.; Guang, X.; Li, A.; Zhao, C.; Yang, W.; et al. Whole-genome sequencing of the endangered bovine species Gayal (Bos frontalis) provides new insights into its genetic features. Sci. Rep. 2016, 6, 19787. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Dogan, H.; Can, H.; Otu, H.H. Whole genome sequence of a Turkish individual. PLoS ONE 2014, 9, e85233. [Google Scholar] [CrossRef]
- Zhou, C.; Zhang, W.; Wen, Q.; Bu, P.; Gao, J.; Wang, G.; Jin, J.; Song, Y.; Sun, X.; Zhang, Y.; et al. Comparative Genomics Reveals the Genetic Mechanisms of Musk Secretion and Adaptive Immunity in Chinese Forest Musk Deer. Genome Biol. Evol. 2019, 11, 1019–1032. [Google Scholar] [CrossRef]
- Akiyama, M. The roles of ABCA12 in epidermal lipid barrier formation and keratinocyte differentiation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2014, 1841, 435–440. [Google Scholar] [CrossRef]
- Jensen, J.M.; Proksch, E. The skin’s barrier. G. Ital. Dermatol. Venereol. Organo Uff. Soc. Ital. Dermatol. Sifilogr. 2009, 144, 689–700. [Google Scholar]
- Kelsell, D.P.; Norgett, E.E.; Unsworth, H.; Teh, M.T.; Cullup, T.; Mein, C.A.; Dopping-Hepenstal, P.J.; Dale, B.A.; Tadini, G.; Fleckman, P.; et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am. J. Hum. Genet. 2005, 76, 794–803. [Google Scholar] [CrossRef]
- Scott, C.A.; Rajpopat, S.; Di, W.L. Harlequin ichthyosis: ABCA12 mutations underlie defective lipid transport, reduced protease regulation and skin-barrier dysfunction. Cell Tissue Res. 2013, 351, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Quan, X.J.; Hassan, B.A. From skin to nerve: Flies, vertebrates and the first helix. Cell. Mol. Life Sci. 2005, 62, 2036–2049. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, M.; Bjorntorp Mark, E.; Brantsing, C.; Brandner, J.M.; Lindahl, A.; Asp, J. Hash4, a novel human achaete-scute homologue found in fetal skin. Genomics 2004, 84, 859–866. [Google Scholar] [CrossRef]
- Rezza, A.; Wang, Z.; Sennett, R.; Qiao, W.; Wang, D.; Heitman, N.; Mok, K.W.; Clavel, C.; Yi, R.; Zandstra, P.; et al. Signaling Networks among Stem Cell Precursors, Transit-Amplifying Progenitors, and their Niche in Developing Hair Follicles. Cell Rep. 2016, 14, 3001–3018. [Google Scholar] [CrossRef]
- Sarasin, A. UVSSA and USP7: New players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med. 2012, 4, 44. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Sasaki, K.; Mitsutake, N.; Matsuse, M.; Shimada, M.; Nardo, T.; Takahashi, Y.; Ohyama, K.; Ito, K.; Mishima, H.; et al. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 2012, 44, 586–592. [Google Scholar] [CrossRef]


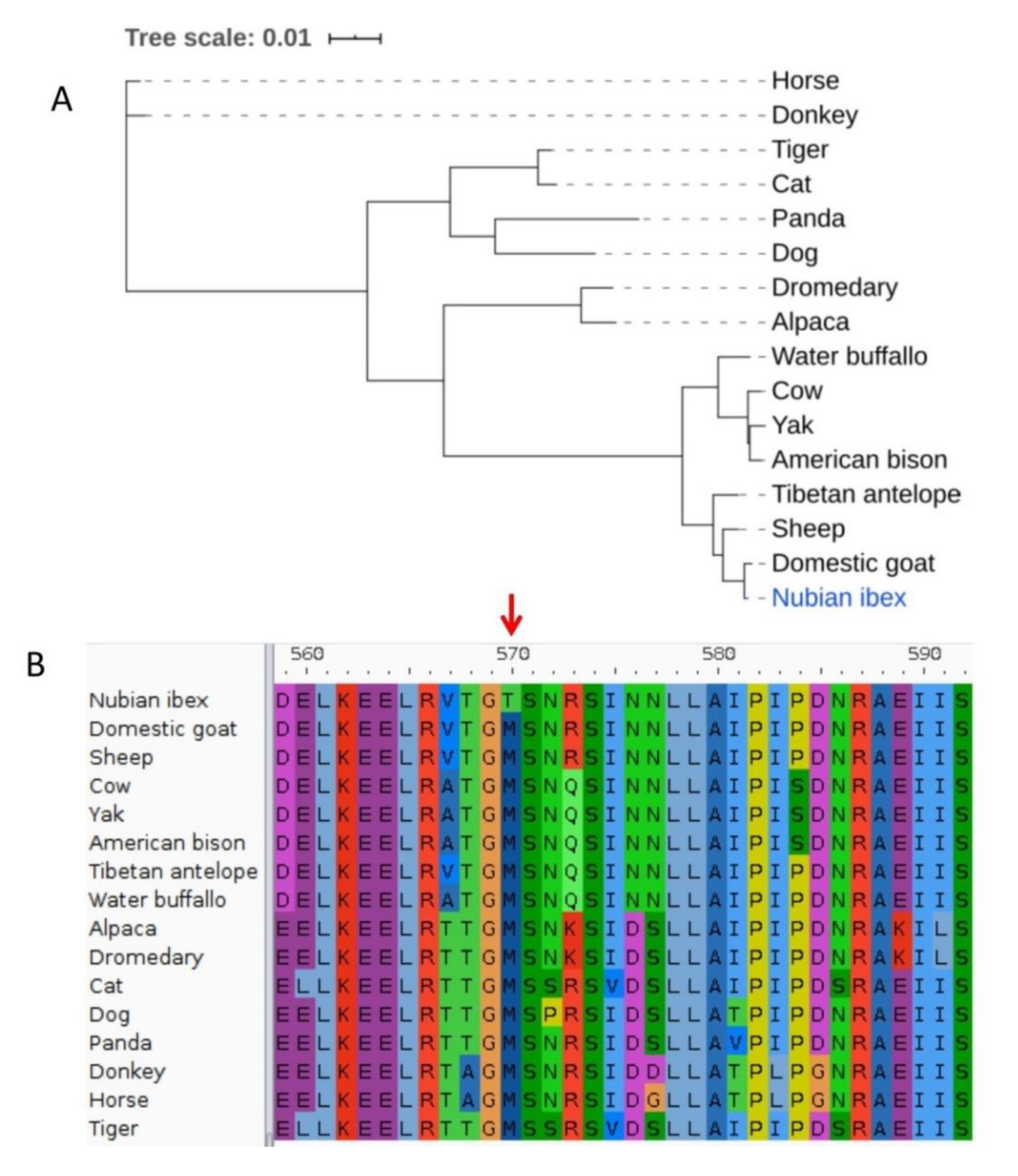{kind=link}
| Ensembl Gene Id | Gene Name | Positively Selected Sites with BEB Posterior Probability > 0.8 | Polyphen-2 Functional Impact Analysis |
|---|---|---|---|
| ENSCHIT00000003090 | Storkhead box 2 | T734V, N835T | Benign |
| ENSCHIT00000004084 | Atpase H+ transporting V1 subunit E2 | M72N | Possibly damaging |
| ENSCHIT00000004434 | Olfactory receptor 2G2-like | F73T | Probably damaging |
| ENSCHIT00000008957 | Serine protease 56 | Q424L, R425G, R436W | Benign, probably damaging, benign |
| ENSCHIT00000010253 | Matrix AAA peptidase interacting protein 1 | T76A, Q93P | Benign |
| ENSCHIT00000012782 | Putative olfactory receptor 52P1 | M67L | Possibly damaging |
| ENSCHIT00000015750 | Prostaglandin I2 synthase | R320H, D411E | Possibly damaging, benign |
| ENSCHIT00000018881 | F-box protein 21 | S603A, E606G, K615E, E620G | Benign |
| K616R | Possibly damaging | ||
| ENSCHIT00000026283 | Zinc finger and SCAN domain containing 23 | P213N | Probably damaging |
| ENSCHIT00000028977 | UV stimulated scaffold protein A | D361G, A517T | Probably damaging benign |
| E99Q | Probably damaging | ||
| ENSCHIT00000030384 | F-box and WD repeat domain containing 2 | L82C | Probably damaging |
| ENSCHIT00000000612 | Multimerin 2 | S214H | Probably damaging |
| ENSCHIT00000015914 | Toll like receptor adaptor molecule 2 | I213N | Benign |
| ENSCHIT00000016318 | Eukaryotic translation initiation factor 2 subunit beta | K83I, K205E | Possibly damaging |
| ENSCHIT00000020934 | LY6/PLAUR domain containing 6B | A7T, F16L | Benign |
| ENSCHIT00000028741 | ATP binding cassette subfamily A member 12 | M570T | Possibly damaging |
| ENSCHIT00000035903 | PATJ crumbs cell polarity complex component | V249I, I1739V | Benign |
| I1738F | Probably damaging | ||
| ENSCHIT00000036547 | Rho gtpase activating protein 42 | I502L, M770T | Benign |
| W773R | Probably damaging | ||
| ENSCHIT00000040177 | Achaete-scute family bhlh transcription factor 4 | L30S | Probably damaging |
| ENSCHIT00000040379 | Olfactory receptor 1P1 | A133T | Benign |
| V135D, H159C | Possibly damaging | ||
| ENSCHIT00000034768 | Tripartite motif containing 16 | D159L, S515L | Benign |
| ENSCHIT00000041152 | Centrosomal protein 112 | K338G | Unknown |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chebii, V.J.; Oyola, S.O.; Kotze, A.; Domelevo Entfellner, J.-B.; Musembi Mutuku, J.; Agaba, M. Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment. Animals 2020, 10, 2181. https://doi.org/10.3390/ani10112181
Chebii VJ, Oyola SO, Kotze A, Domelevo Entfellner J-B, Musembi Mutuku J, Agaba M. Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment. Animals. 2020; 10(11):2181. https://doi.org/10.3390/ani10112181
Chicago/Turabian StyleChebii, Vivien J., Samuel O. Oyola, Antoinette Kotze, Jean-Baka Domelevo Entfellner, J. Musembi Mutuku, and Morris Agaba. 2020. "Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment" Animals 10, no. 11: 2181. https://doi.org/10.3390/ani10112181
APA StyleChebii, V. J., Oyola, S. O., Kotze, A., Domelevo Entfellner, J.-B., Musembi Mutuku, J., & Agaba, M. (2020). Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment. Animals, 10(11), 2181. https://doi.org/10.3390/ani10112181