Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction

 ,
, {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Models of HFrEF
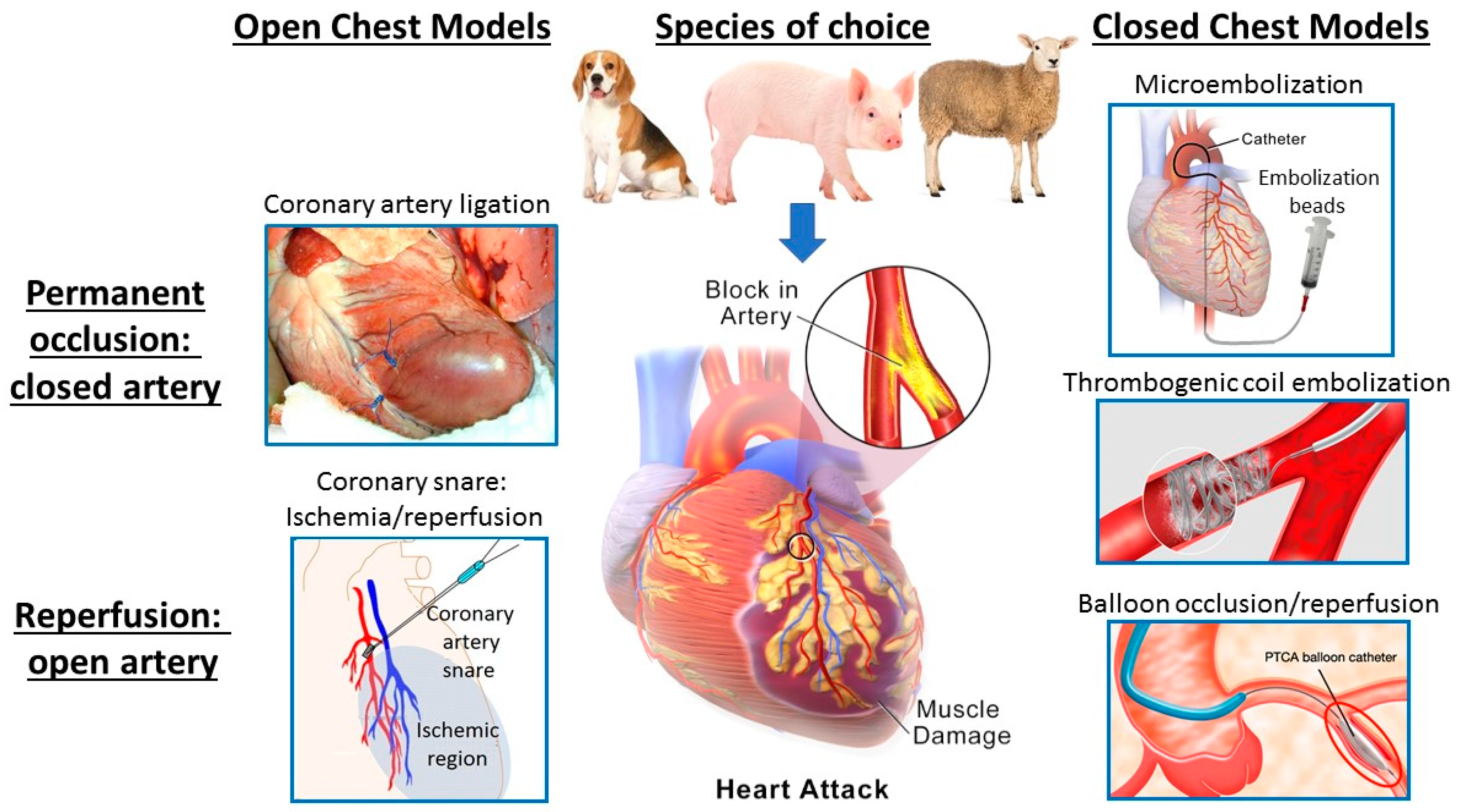
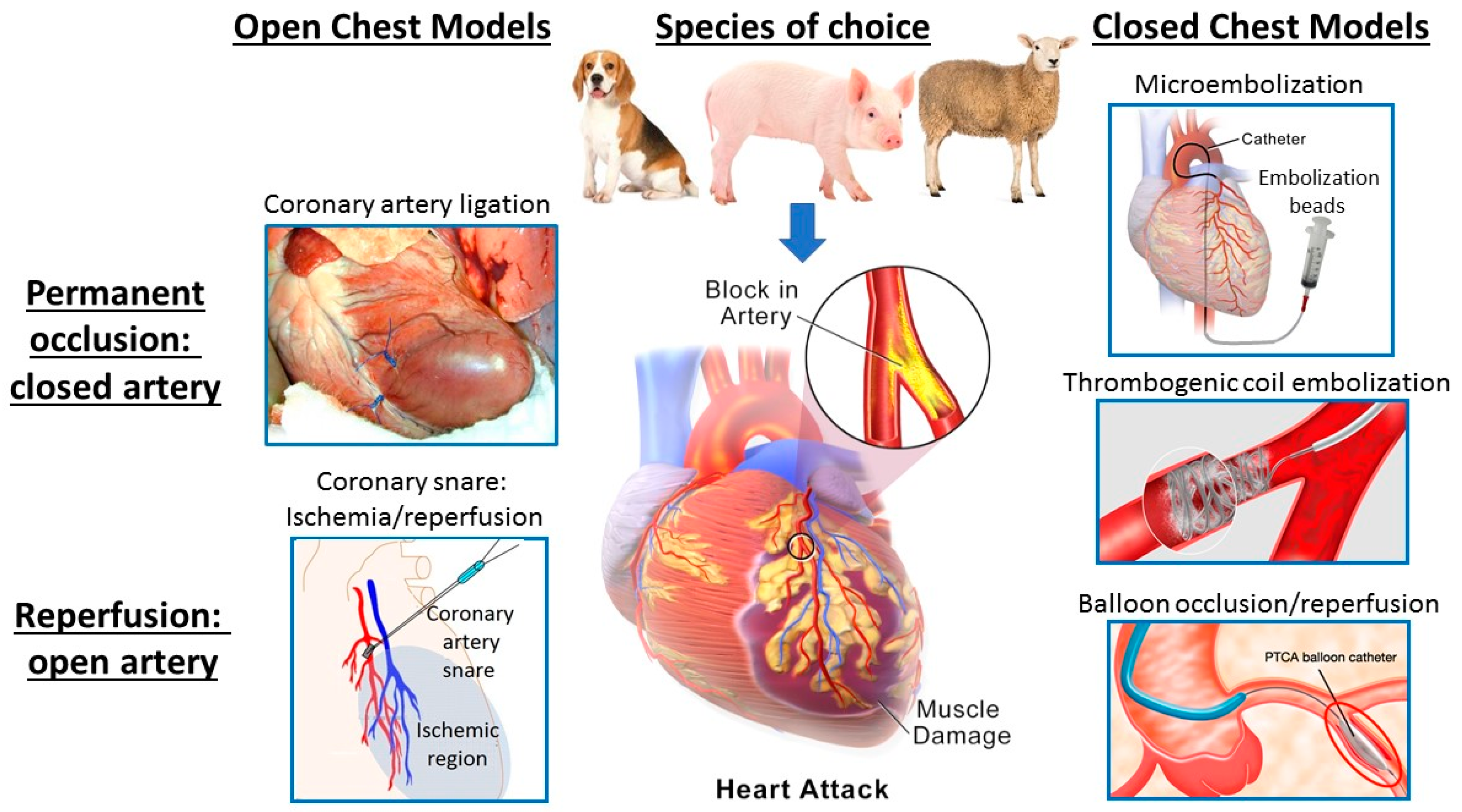
2.1. Models of Acute Myocardial Ischemia/Infarction
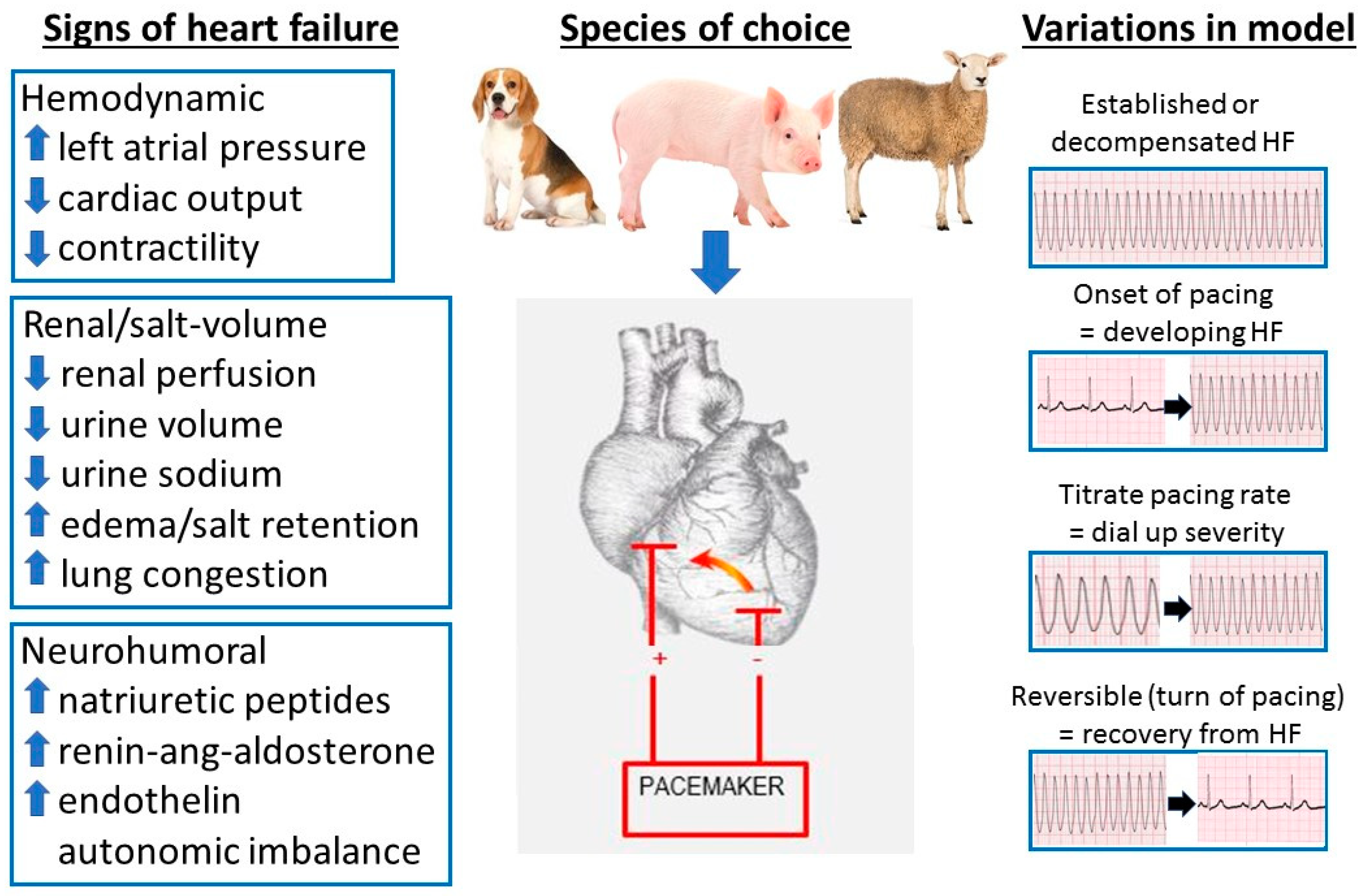
2.2. Pacing Models of HF
3. Models of HFpEF
Pressure Overload Models of HFpEF
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal Models of Heart Failure. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Jones, D.M.; Larson, M.G.; Leip, E.P.; Beiser, A.; D’Agostino, R.B.; Kannel, W.B.; Murabito, J.M.; Vasan, R.S.; Benjamin, E.J.; Levy, D. Lifetime Risk for Developing Congestive Heart Failure. Circulation 2002, 106, 3068–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.J. Heart Disease and Stroke Statistics—2017 Update: A report from the AHA. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kenchaiah, S.; Larson, M.G.; Benjamin, E.J.; Kupka, M.J.; Ho, K.K.; Murabito, J.M.; Vasan, R.S. Long-Term Trends in the Incidence of and Survival with Heart Failure. N. Engl. J. Med. 2002, 347, 1397–1402. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Foneseca, C.; Gomez-Sanchez, M.A.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in Prevalence and Outcome of Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, R.S.; Tu, J.V.; Lee, D.S.; Austin, P.C.; Fang, J.; Haouzi, A.; Gong, Y.; Liu, P.P. Outcome of Heart Failure with Preserved Ejection Fraction in a Population-Based Study. N. Engl. J. Med. 2006, 355, 260–269. [Google Scholar] [CrossRef] [Green Version]
- MAGGIC Collaborative Group. The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: An individual patient data meta-analysis. Eur. Heart J. 2012, 33, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.S.; Gamble, G.D.; Ling, L.H.; Sim, D.; Leong, K.T.G.; Yeo, P.S.; Ong, H.Y.; Jaufeerally, F.; Ng, T.P.; Cameron, V.A.; et al. Mortality associated with heart failure with preserved vs. reduced ejection fraction in a prospective international multi-ethnic cohort study. Eur. Heart J. 2018, 39, 1770–1780. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.L.; Chan, F.T.; A Nabeebaccus, A.; Shah, A.M.; McDonagh, T.; O Okonko, D.; Ayis, S. Drug treatment effects on outcomes in heart failure with preserved ejection fraction: A systematic review and meta-analysis. Heart 2017, 104, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.; Houstis, N.; Rosenzweig, A. Why Don’t We Have Proven Treatments for HFpEF? Circ. Res. 2017, 120, 1243–1245. [Google Scholar] [CrossRef]
- Conceição, G.; Heinonen, I.; Lourenço, A.P.; Duncker, D.J.; Falcão-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Heart J. 2016, 24, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Fish, K.; Bonnet, G.; Santos-Gallego, C.G.; Leonardson, L.; Hajjar, R.J.; Ishikawa, K. Echocardiographic and hemodynamic assessment for predicting early clinical events in severe acute mitral regurgitation. Int. J. Cardiovasc. Imag. 2017, 34, 171–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessier, D.; Lajos, P.; Braunberger, E.; Pouchelon, J.-L.; Carpentier, A.; Chachques, J.C.; Chetboul, V. Induction of Chronic Cardiac Insufficiency by Arteriovenous Fistula and Doxorubicin Administration. J. Card. Surg. 2003, 18, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Goetzenich, A.; Hatam, N.; Zernecke, A.; Weber, C.; Czarnotta, T.; Autschbach, R.; Christiansen, S. Alteration of Matrix Metalloproteinases in Selective Left Ventricular Adriamycin-Induced Cardiomyopathy in the Pig. J. Heart Lung Transplant. 2009, 28, 1087–1093. [Google Scholar] [CrossRef]
- Carbone, A.; Psaltis, P.J.; Nelson, A.J.; Metcalf, R.; Richardson, J.D.; Weightman, M.J.; Thomas, A.; Finnie, J.W.; Young, G.D.; Worthley, S.G. Dietary Omega-3 Supplementation Exacerbates Left Ventricular Dysfunction in an Ovine Model of Anthracycline-Induced Cardiotoxicity. J. Card. Fail. 2012, 18, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Spannbauer, A.; Traxler, D.; Zlabinger, K.; Gugerell, A.; Winkler, J.; Mester-Tonczar, J.; Lukovic, D.; Müller, C.; Riesenhuber, M.; Pavo, N.; et al. Large Animal Models of Heart Failure With Reduced Ejection Fraction (HFrEF). Front. Cardiovasc. Med. 2019, 6, 117. [Google Scholar] [CrossRef] [Green Version]
- Reimer, K.; Jennings, R.B. The “wavefront phenomenon” of myocardial ischemic cell death. II. Transmural progression of necrosis within the framework of ischemic bed size (myocardium at risk) and collateral flow. Lab. Investig. 1979, 40, 633–644. [Google Scholar]
- Reimer, K.; E Lowe, J.; Rasmussen, M.M.; Jennings, R.B. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977, 56, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.J.; Charles, C.J.; Sands, J.; Richards, A.M.; Bridgman, P.G.; Gooneratne, R.; Ikram, H. An ovine model of acute myocardial infarction and chronic left ventricular dysfunction. Angiology 1997, 48, 679–688. [Google Scholar] [CrossRef]
- Charles, C.J.; Elliott, J.M.; Nicholls, M.G.; Rademaker, M.T.; Richards, A.M. Myocardial infarction with and without reperfusion in sheep: Early cardiac and neurohumoral changes. Clin. Sci. 2000, 98, 703–711. [Google Scholar] [CrossRef]
- Rademaker, M.T.; Cameron, V.A.; Charles, C.J.; Espiner, E.A.; Nicholls, M.; Pemberton, C.J.; Richards, A.M. Neurohormones in an ovine model of compensated postinfarction left ventricular dysfunction. Am. J. Physiol. Circ. Physiol. 2000, 278, H731–H740. [Google Scholar] [CrossRef]
- Jardine, D.; Charles, C.J.; Ashton, R.K.; Bennett, S.I.; Whitehead, M.; Frampton, C.M.; Nicholls, M.G. Increased cardiac sympathetic nerve activity following acute myocardial infarction in a sheep model. J. Physiol. 2005, 565, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Charles, C.; Jardine, D.; Richards, A. Cardiac sympathetic nerve activity and ventricular fibrillation during acute myocardial infarction in ambulent sheep. J. Mol. Cell. Cardiol. 2007, 42, S8. [Google Scholar] [CrossRef]
- Monreal, G.; A Gerhardt, M.; Kambara, A.; Abrishamchian, A.R.; A Bauer, J.; Goldstein, A.H. Selective microembolization of the circumflex coronary artery in an ovine model: Dilated, ischemic cardiomyopathy and left ventricular dysfunction. J. Card. Fail. 2004, 10, 174–183. [Google Scholar] [CrossRef]
- O’Rourke, B.; Kass, D.A.; Tomaselli, G.F.; Kääb, S.; Tunin, R.; Marbaán, E. Mechanisms of Altered Excitation-Contraction Coupling in Canine Tachycardia-Induced Heart Failure, I. Circ. Res. 1999, 84, 562–570. [Google Scholar] [CrossRef] [Green Version]
- McMahon, W.S.; Mukherjee, R.; Gillette, P.C.; A Crawford, F.; Spinale, F.G. Right and left ventricular geometry and myocyte contractile processes with dilated cardiomyopathy: Myocyte growth and beta-adrenergic responsiveness. Cardiovasc. Res. 1996, 31, 314–323. [Google Scholar]
- Fitzpatrick, M.A.; Nicholls, M.G.; Espiner, E.A.; Ikram, H.; Bagshaw, P.; Yandle, T.G. Neurohumoral changes during onset and offset of ovine heart failure: Role of ANP. Am. J. Physiol. Circ. Physiol. 1989, 256, H1052–H1059. [Google Scholar] [CrossRef]
- Perreault, C.L.; Shannon, R.P.; Komamura, K.; Vatner, S.F.; Morgan, J.P. Abnormalities in intracellular calcium regulation and contractile function in myocardium from dogs with pacing-induced heart failure. J. Clin. Investig. 1992, 89, 932–938. [Google Scholar] [CrossRef] [Green Version]
- Spinale, F.G.; Tomita, M.; Zellner, J.L.; Cook, J.C.; Crawford, F.A.; Zile, M.R. Collagen remodeling and changes in LV function during development and recovery from supraventricular tachycardia. Am. J. Physiol. Circ. Physiol. 1991, 261, H308–H318. [Google Scholar] [CrossRef]
- Rademaker, M.T.; Charles, C.J.; Espiner, E.A.; Frampton, C.M.; Nicholls, M.G.; Richards, A.M. Natriuretic peptide responses to acute and chronic ventricular pacing in sheep. Am. J. Physiol. Circ. Physiol. 1996, 270, H594–H602. [Google Scholar] [CrossRef]
- Rademaker, M.T.; Ellmers, L.J.; Charles, C.J.; Richards, M. Urocortin 2 protects heart and kidney structure and function in an ovine model of acute decompensated heart failure: Comparison with dobutamine. Int. J. Cardiol. 2015, 197, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, M.T.; Charles, C.J.; Richards, A.M. Urocortin 1 administration from onset of rapid left ventricular pacing represses progression to overt heart failure. Am. J. Physiol. Circ. Physiol. 2007, 293, H1536–H1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, C.J.; Prickett, T.C.R.; Espiner, E.A.; Rademaker, M.T.; Richards, A.M.; Yandle, T.G. Regional sampling and the effects of experimental heart failure in sheep: Differential responses in A, B and C-type natriuretic peptides. Peptides 2006, 27, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Ramchandra, R.; Hood, S.G.; Frithiof, R.; McKinley, M.J.; May, C.N. The role of the paraventricular nucleus of the hypothalamus in the regulation of cardiac and renal sympathetic nerve activity in conscious normal and heart failure sheep. J. Physiol. 2012, 591, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Reiter, U.; Reiter, G.; Manninger, M.; Adelsmayr, G.; Schipke, J.; Alogna, A.; Rajces, A.; Stalder, A.; Greiser, A.; Mühlfeld, C.; et al. Early-stage heart failure with preserved ejection fraction in the pig: A cardiovascular magnetic resonance study. J. Cardiovasc. Magn. Reson. 2016, 18, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzl, M.; Hamdani, N.; Seiler, S.; Alogna, A.; Manninger, M.; Reilly, S.; Zirngast, B.; Kirsch, A.H.; Steendijk, P.; Verderber, J.; et al. A porcine model of hypertensive cardiomyopathy: Implications for heart failure with preserved ejection fraction. Am. J. Physiol. Circ. Physiol. 2015, 309, H1407–H1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorop, O.; Heinonen, I.; Van Kranenburg, M.; Van De Wouw, J.; De Beer, V.J.; Nguyen, I.T.N.; Octavia, Y.; Van Duin, R.W.B.; Stam, K.; Van Geuns, R.-J.; et al. Multiple common comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc. Res. 2018, 114, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Charles, C.J.; Kaaja, R.J.; Espiner, E.A.; Nicholls, M.G.; Pemberton, C.J.; Richards, A.M.; Yandle, T.G. Natriuretic Peptides in Sheep with Pressure Overload Left Ventricular Hypertrophy. Clin. Exp. Hypertens. 1996, 18, 1051–1071. [Google Scholar] [CrossRef]
- Xiong, Q.; Zhang, P.; Guo, J.; Swingen, C.; Jang, A.; Zhang, J. Myocardial ATP hydrolysis rates in vivo: A porcine model of pressure overload-induced hypertrophy. Am. J. Physiol. Circ. Physiol. 2015, 309, H450–H458. [Google Scholar] [CrossRef] [Green Version]
- Hiemstra, J.A.; Liu, S.; Ahlman, M.A.; Schuleri, K.H.; Lardo, A.C.; Baines, C.P.; Dellsperger, K.C.; Bluemke, D.A.; Emter, C.A. A new twist on an old idea: A two-dimensional speckle tracking assessment of cyclosporine as a therapeutic alternative for heart failure with preserved ejection fraction. Physiol. Rep. 2013, 1, 10074. [Google Scholar] [CrossRef] [PubMed]
- Hamdani, N.; Bishu, K.G.; Von Frieling-Salewsky, M.; Redfield, M.M.; Linke, W.A. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc. Res. 2012, 97, 464–471. [Google Scholar] [CrossRef] [Green Version]
- Yarbrough, W.M.; Mukherjee, R.; Stroud, R.E.; Rivers, W.T.; Oelsen, J.M.; Dixon, J.A.; Eckhouse, S.R.; Ikonomidis, J.S.; Zile, M.R.; Spinale, F.G. Progressive induction of left ventricular pressure overload in a large animal model elicits myocardial remodeling and a unique matrix signature. J. Thorac. Cardiovasc. Surg. 2012, 143, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, C.J.; Lee, P.; Li, R.R.; Yeung, T.; Mazlan, S.M.I.; Tay, Z.W.; Abdurrachim, D.; Teo, X.Q.; Wang, W.; De Kleijn, D.P.; et al. A porcine model of heart failure with preserved ejection fraction: Magnetic resonance imaging and metabolic energetics. ESC Heart Fail. 2019, 7, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, A.P.; Leite-Moreira, A.; Balligand, J.-L.; Bauersachs, J.; Dawson, D.; De Boer, R.A.; De Windt, L.J.; Falcão-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2017, 20, 216–227. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charles, C.J.; Rademaker, M.T.; Scott, N.J.A.; Richards, A.M. Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction. Animals 2020, 10, 1906. https://doi.org/10.3390/ani10101906
Charles CJ, Rademaker MT, Scott NJA, Richards AM. Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction. Animals. 2020; 10(10):1906. https://doi.org/10.3390/ani10101906
Chicago/Turabian StyleCharles, Christopher J., Miriam T. Rademaker, Nicola J. A. Scott, and A. Mark Richards. 2020. "Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction" Animals 10, no. 10: 1906. https://doi.org/10.3390/ani10101906
APA StyleCharles, C. J., Rademaker, M. T., Scott, N. J. A., & Richards, A. M. (2020). Large Animal Models of Heart Failure: Reduced vs. Preserved Ejection Fraction. Animals, 10(10), 1906. https://doi.org/10.3390/ani10101906