The Rumen Specific Bacteriome in Dry Dairy Cows and Its Possible Relationship with Phenotypes

 ,
,
Abstract
:Simple Summary
Abstract
1. Background
2. Methods
2.1. Animal Sources and Sampling
2.2. DNA Extraction and Next-Generation Sequencing
2.3. Bioinformatics
3. Results
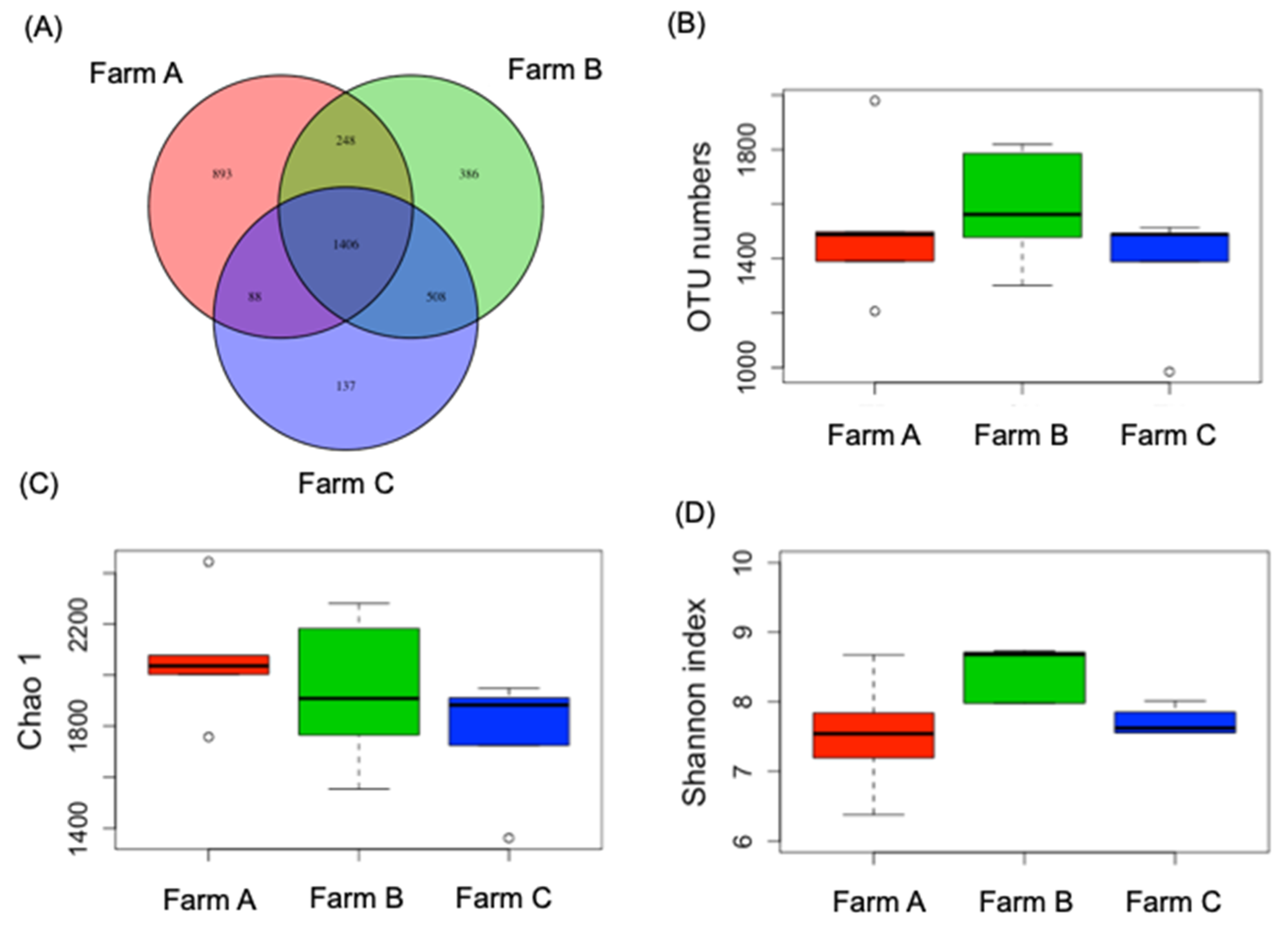
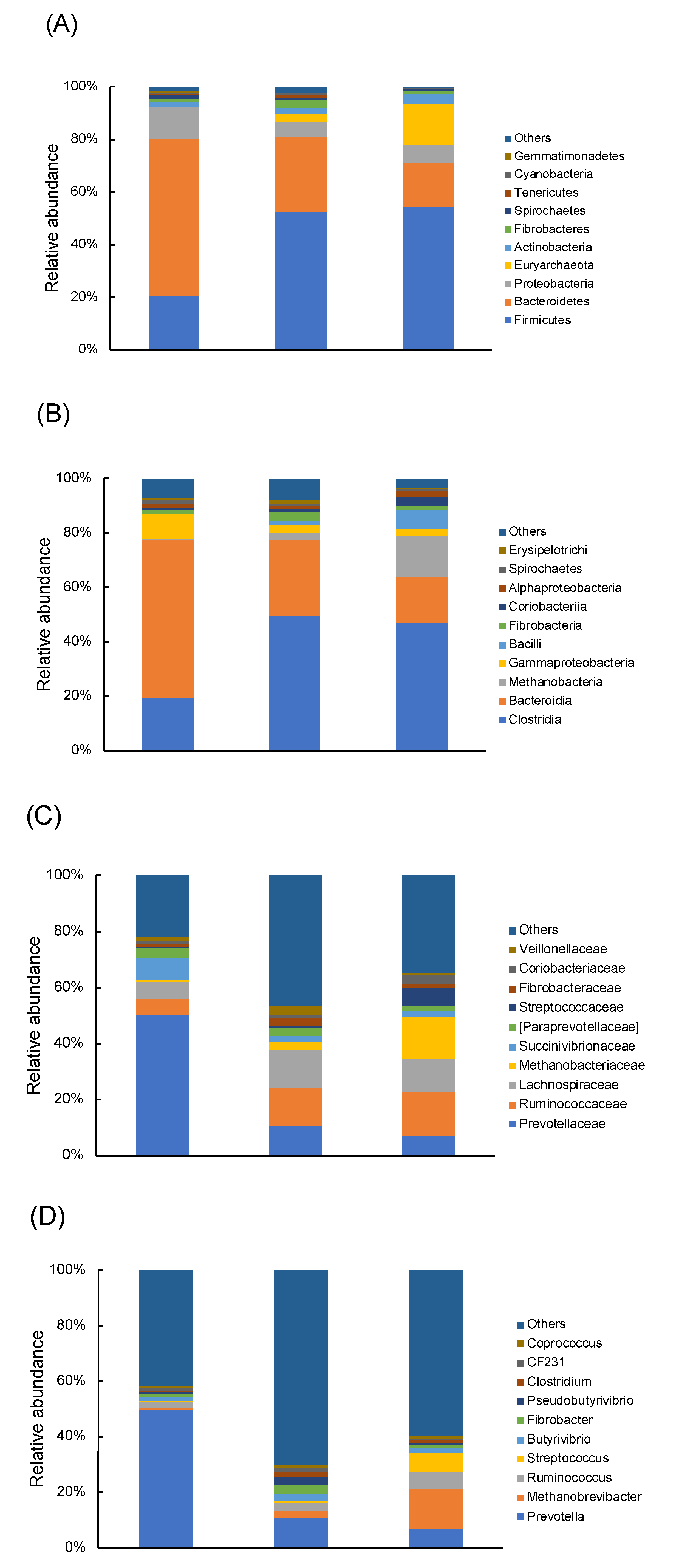
3.1. Proportion of Rumen Bacterial Communities in Dry Cows Was Diverse Across Regional Farms
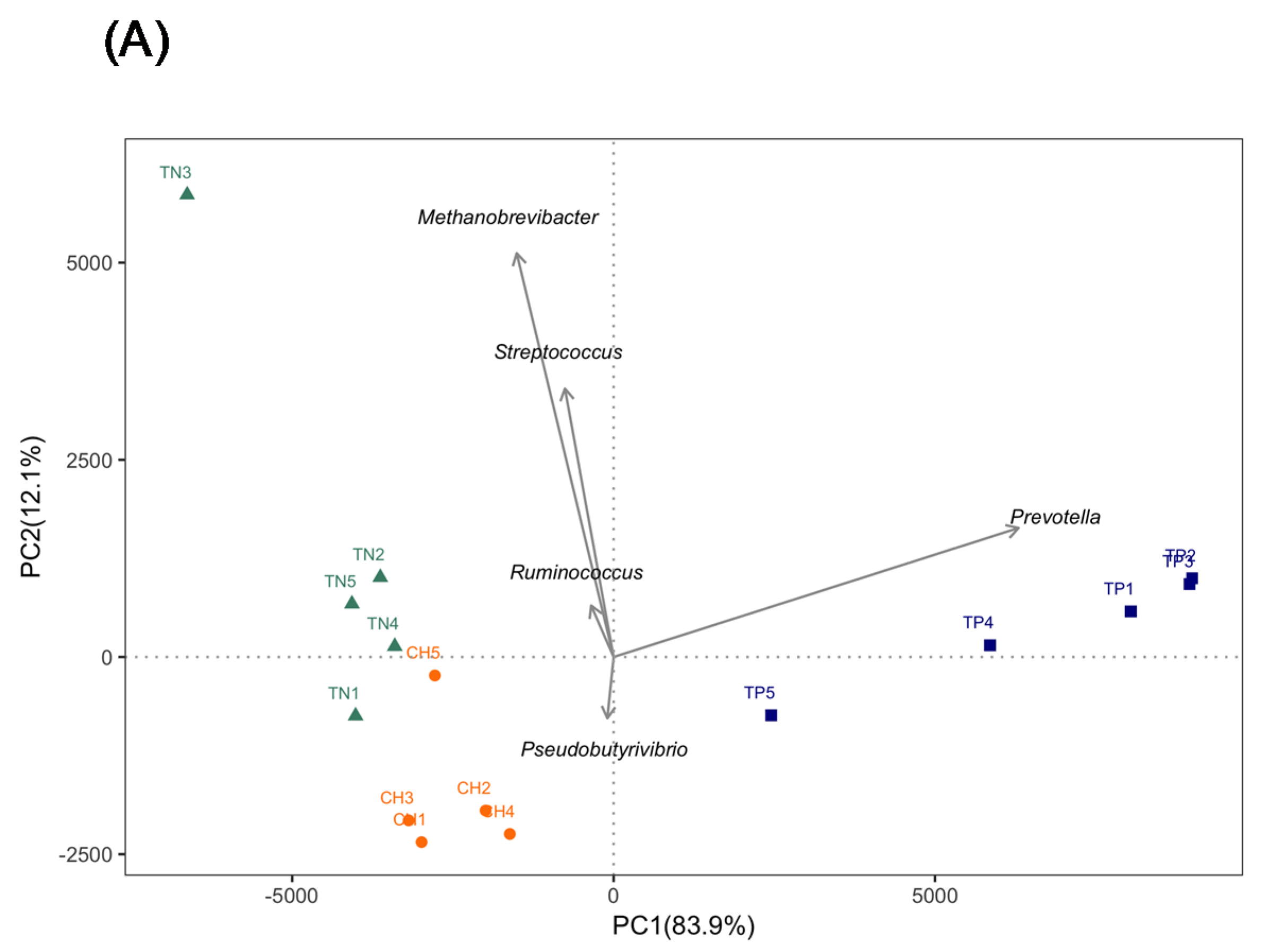
3.2. Dry Cows from Regional Farms Contained Clearly Recognizable Rumen Microbiota
3.3. LEfSe Analysis Identified Biomarkers of Rumen Microbiota from Regional Farms
3.4. Diet Composition Affected the Network of Co-Occurring Predominant Bacteria at the Genus Level in Rumen
3.5. Functional Prediction Revealed Similar Physiological Functions of Rumen Microbiota in Dry Cows across Regional Farms
4. Discussion
5. Conclusions
6. Declarations
6.1. Consent for Publication
6.2. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | abundance-based coverage estimators |
| CF | crude fiber |
| CP | crude protein |
| KEGG | Kyoto Encyclopedia of Genes and Genome |
| LDA | linear discriminant analysis |
| OTU | operational taxonomic unit |
| PCA | principal components analysis |
| PICRUSt | phylogenetic investigation of communities by reconstruction of unobserved states |
| rRNA | ribosomal RNA |
| SCFA | short-chain fatty acid |
References
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H.; Global Rumen Census Collaborators. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Guan, L.L. Understanding host-microbial interactions in rumen: Searching the best opportunity for microbiota manipulation. J. Anim. Sci. Biotechnol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Houtert, M.F.J. The production and metabolism of volatile fatty acids by ruminants fed roughages: A review. Anim. Feed Sci. Technol. 1993, 43, 189–225. [Google Scholar] [CrossRef]
- Bauman, D.E.; Harvatine, K.J.; Lock, A.L. Nutrigenomics, rumen-derived bioactive fatty acids, and the regulation of milk fat synthesis. Annu. Rev. Nutr. 2011, 31, 299–319. [Google Scholar] [CrossRef]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome-metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2016, 18, 525–541. [Google Scholar] [CrossRef]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Zhao, C.; Hu, P.; Chen, H.; Liu, Z.; Liu, G.; Wang, Z. The correlation between composition of the bacterial community and concentration of volatile fatty acids in the rumen during the transition period and ketosis in dairy cows. Appl. Environ. Microbiol. 2012, 78, 2386–2392. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, T.G.; Titgemeyer, E.C. Ruminal acidosis in beef cattle: The current microbiological and nutritional outlook. J. Dairy Sci. 2007, 90, E17–E38. [Google Scholar] [CrossRef] [Green Version]
- Hook, S.E.; Steele, M.A.; Northwood, K.S.; Dijkstra, J.; France, J.; Wright, A.D.; McBride, B.W. Impact of subacute ruminal acidosis (SARA) adaptation and recovery on the density and diversity of bacteria in the rumen of dairy cows. FEMS Microbiol. Ecol. 2011, 78, 275–284. [Google Scholar] [CrossRef] [Green Version]
- McCann, J.C.; Luan, S.; Cardoso, F.C.; Derakhshani, H.; Khafipour, E.; Loor, J.J. Induction of subacute ruminal acidosis affects the ruminal microbiome and epithelium. Front. Microbiol. 2016, 7, 701. [Google Scholar] [CrossRef] [Green Version]
- Weimer, P.J. Redundancy, resilience, and host specificity of the ruminal microbiota: Implications for engineering improved ruminal fermentations. Front. Microbiol. 2015, 6, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Hou, Q.; Wang, Y.; Ma, H.; Liu, Y.; Zhao, F.; Li, J.; Kwok, L.Y.; Yu, J.; Sun, Z.; et al. Analysis of the gut microbial diversity of dairy cows during peak lactation by PacBio single-molecule real-time (SMRT) sequencing. Curr. Microbiol. 2018, 275, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Sun, H.; Wu, X.; Guan, L.L.; Liu, J. Assessment of rumen microbiota from a large dairy cattle cohort reveals the pan and core bacteriomes contributing to varied phenotypes. Appl. Environ. Microbiol. 2018, 84, e00970-18. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2010, 26, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Gevers, D.; Earl, A.M.; Feldgarden, M.; Ward, D.V.; Giannoukos, G.; Ciulla, D.; Tabbaa, D.; Highlander, S.K.; Sodergren, E.; et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011, 21, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittingerb, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.H. Evolution and measurement of species diversity. Taxon 1972, 21, 213–251. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 2012, 7, e33306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Guan, L.L. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 2017, 83, e00061-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotechnol. 2007, 75, 165–174. [Google Scholar] [CrossRef]
- Wallace, R.J.; Chaudhary, L.C.; McKain, N.; McEwan, N.R.; Richardson, A.J.; Vercoe, P.E.; Walker, N.D.; Paillard, D. Clostridium proteoclasticum: A ruminal bacterium that forms stearic acid from linoleic acid. FEMS Microbiol. Lett. 2006, 265, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Emerson, E.L.; Weimer, P.J. Fermentation of model hemicelluloses by Prevotella strains and Butyrivibrio fibrisolvens in pure culture and in ruminal enrichment cultures. Appl. Microbiol. Biotechnol. 2017, 101, 4269–4278. [Google Scholar] [CrossRef]
- Walker, A.W.; Duncan, S.H.; McWilliam Leitch, E.C.; Child, M.W.; Flint, H.J. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl. Environ. Microbiol. 2005, 71, 3692–3700. [Google Scholar] [CrossRef] [Green Version]
- Mrázek, J.; Tepšič, K.; Kopečný, J.; Avguštin, G. Diet-dependent shifts in ruminal butyrate-producing bacteria. Folia Microbiol. 2006, 51, 294–298. [Google Scholar] [CrossRef]
- Zhong, Y.; Xue, M.; Liu, J. Composition of rumen bacterial community in dairy cows with different levels of somatic cell counts. Front. Microbiol. 2018, 9, 3217. [Google Scholar] [CrossRef] [Green Version]
- Indugu, N.; Vecchiarelli, B.; Baker, L.D.; Ferguson, J.D.; Vanamala, J.K.P.; Pitta, D.W. Comparison of rumen bacterial communities in dairy herds of different production. BMC Microbiol. 2017, 17, 190. [Google Scholar] [CrossRef]
- Fondevila, M.; Dehority, B.A. Interactions between Fibrobacter succinogenes, Prevotella ruminicola, and Ruminococcus flavefaciens in the digestion of cellulose from forages. J. Anim. Sci. 1996, 74, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Sato, T.; Yamada, T. Metabolic pathways for cytotoxic end product formation from glutamate-and aspartate-containing peptides by Porphyromonas gingivalis. J. Bacteriol. 2000, 182, 4704–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugaut, M. Occurrence, absorption and metabolism of short chain fatty acids in the digestive tract of mammals. Comp. Biochem. Physiol. B 1987, 86, 439–472. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [Green Version]
- Chilliard, Y.; Ferlay, A.; Mansbridge, R.M.; Doreau, M.; Agabriel, J.; Givens, I. Ruminant milk fat plasticity: Nutritional control of saturated, polyunsaturated, trans and conjugated fatty acids. Ann. Zootech. 2000, 49, 181–205. [Google Scholar] [CrossRef] [Green Version]
- Grilli, D.J.; Cerón, M.E.; Paez, S.; Egea, V.; Schnittger, L.; Cravero, S.; Escudero, M.S.; Allegretti, L.; Arenas, G.N. Isolation of Pseudobutyrivibrio ruminis and Pseudobutyrivibrio xylanivorans from rumen of Creole goats fed native forage diet. Folia Microbiol. 2013, 58, 367–373. [Google Scholar] [CrossRef]
- Danielsson, R.; Schnürer, A.; Arthurson, V.; Bertilsson, J. Methanogenic population and CH4 production in Swedish dairy cows fed different levels of forage. Appl. Environ. Microbiol. 2012, 78, 6172–6179. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, R.; Dicksved, J.; Sun, L.; Gonda, H.; Müller, B.; Schnürer, A.; Bertilsson, J. Methane production in dairy cows correlates with rumen methanogenic and bacterial community structure. Front. Microbiol. 2017, 8, 1e15. [Google Scholar] [CrossRef]
- King, E.E.; Smith, R.P.; St-Pierre, B.; Wright, D.G. Differences in the rumen methanogen populations of lactating jersey and holstein dairy cows under the same diet regimen. Appl. Environ. Microbiol. 2011, 77, 5682–5687. [Google Scholar] [CrossRef] [Green Version]
- De Menezes, A.B.; Lewis, E.; O’Donovan, M.; O’Neill, B.F.; Clipson, N.; Doyle, E.M. Microbiome analysis of dairy cows fed pasture or total mixed ration diets. FEMS Microbiol. Ecol. 2011, 78, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Cunha, F.; Jeon, S.J.; Daetz, R.; Vieira-Neto, A.; Laporta, J.; Jeong, K.C.; Barbet, A.F.; Risco, C.A.; Galvão, K.N. Quantifying known and emerging uterine pathogens, and evaluating their association with metritis and fever in dairy cows. Theriogenology 2018, 114, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Huang, W.; Hou, Q.; Kwok, L.-Y.; Sun, Z.; Ma, H.; Lee, Y.-K.; Zhang, H. The effects of probiotics administration on the milk production, milk components and fecal bacteria microbiota of dairy cows. Sci. Bull. 2017, 62, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Uyeno, Y.; Shigemori, S.; Shimosato, T. Effect of probiotics/prebiotics on cattle health and productivity. Microbes Environ. 2015, 30, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizrahi, I.; Jami, E. Review: The compositional variation of the rumen microbiome and its effect on host performance and methane emission. Animal 2018, 12, 220–232. [Google Scholar] [CrossRef] [Green Version]
- Hart, E.H.; Creevey, C.J.; Hitch, T.; Kingston-Smith, A.H. Meta-proteomics of rumen microbiota indicates niche compartmentalisation and functional dominance in a limited number of metabolic pathways between abundant bacteria. Sci. Rep. 2018, 8, 10504. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J. The proteolytic systems of ruminal microorganisms. Ann. Zootech. 1996, 45, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Rubino, F.; Carberry, C.; Waters, S.M.; Kenny, D.; McCabe, M.S.; Creevey, C.J. Divergent functional isoforms drive niche specialisation for nutrient acquisition and use in rumen microbiome. ISME J. 2017, 11, 1510. [Google Scholar] [CrossRef] [Green Version]
- Koike, S.; Kobayashi, Y. Fibrolytic rumen bacteria: Their ecology and functions asian-aust. J. Anim. Sci. 2009, 22, 131–138. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm A | |
| Major ingredient | Dry cow concentrate, Bermuda grass |
| F:C (DM) | 85.7:14:3 |
| Crude protein (% DM) | 10.71 |
| Crude fiber (% DM) | 60.90 |
| Calcium (% DM) | 0.60 |
| Phosphorus (% DM) | 0.25 |
| Farm B | |
| Major ingredient | Dry cow concentrate, Bermuda grass, Pennisetum grass |
| F:C (DM) | 88.8:11.2 |
| Crude protein (% DM) | 9.92 |
| Crude fiber (% DM) | 66.79 |
| Calcium (% DM) | 0.66 |
| Phosphorus (% DM) | 0.34 |
| Farm C | |
| Major ingredient | Dry cow concentrate, Bermuda grass, corn silage, oat grass |
| F:C ratio | 67.5:32.5 |
| Crude protein (% DM) | 13.20 |
| Crude fiber (% DM) | 43.08 |
| Calcium (% DM) | 0.45 |
| Phosphorus (% DM) | 0.39 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, S.-T.; Ho, S.-T.; Tu, P.-W.; Li, K.-Y.; Kuo, Y.-L.; Shiu, J.-S.; Wang, S.-Y.; Chen, M.-J. The Rumen Specific Bacteriome in Dry Dairy Cows and Its Possible Relationship with Phenotypes. Animals 2020, 10, 1791. https://doi.org/10.3390/ani10101791
Chuang S-T, Ho S-T, Tu P-W, Li K-Y, Kuo Y-L, Shiu J-S, Wang S-Y, Chen M-J. The Rumen Specific Bacteriome in Dry Dairy Cows and Its Possible Relationship with Phenotypes. Animals. 2020; 10(10):1791. https://doi.org/10.3390/ani10101791
Chicago/Turabian StyleChuang, Shih-Te, Shang-Tse Ho, Po-Wen Tu, Kuan-Yi Li, Yu-Lun Kuo, Jia-Shian Shiu, Sheng-Yao Wang, and Ming-Ju Chen. 2020. "The Rumen Specific Bacteriome in Dry Dairy Cows and Its Possible Relationship with Phenotypes" Animals 10, no. 10: 1791. https://doi.org/10.3390/ani10101791
APA StyleChuang, S.-T., Ho, S.-T., Tu, P.-W., Li, K.-Y., Kuo, Y.-L., Shiu, J.-S., Wang, S.-Y., & Chen, M.-J. (2020). The Rumen Specific Bacteriome in Dry Dairy Cows and Its Possible Relationship with Phenotypes. Animals, 10(10), 1791. https://doi.org/10.3390/ani10101791