Identification of Novel lncRNAs Differentially Expressed in Placentas of Chinese Ningqiang Pony and Yili Horse Breeds

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Samples
2.3. RNA Extraction, Library Preparation, and Sequencing
2.4. Trimming and Mapping of Reads
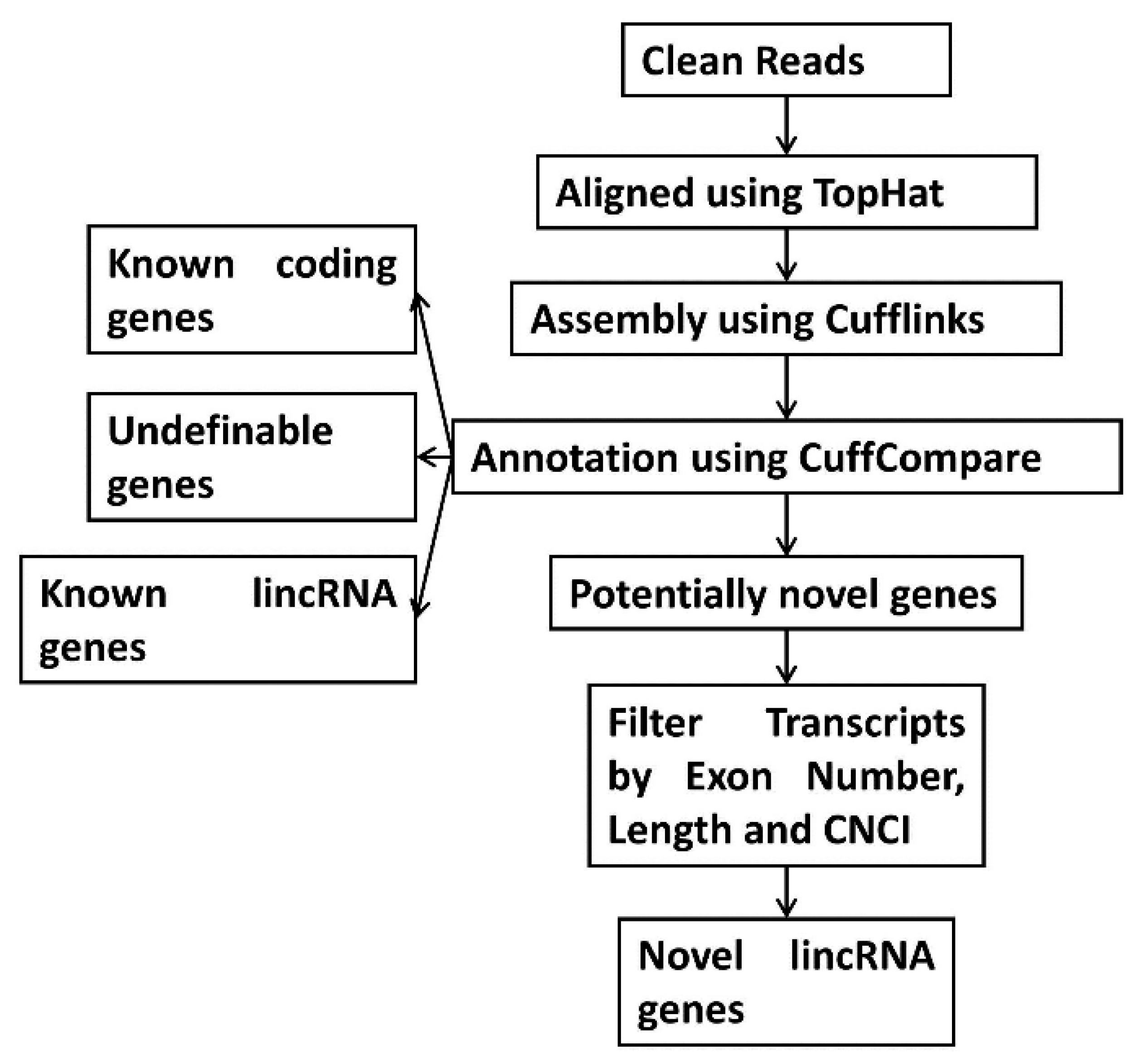
2.5. Step-Wise Filtering for Transcripts
2.6. Identification of lncRNAs from the Assembled Transcripts
2.7. Quantification of lncRNA Expression
2.8. Prediction of Target Genes
2.9. Gene Ontology (GO) Analysis
2.10. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
3.1. Overview of RNA-Seq Data
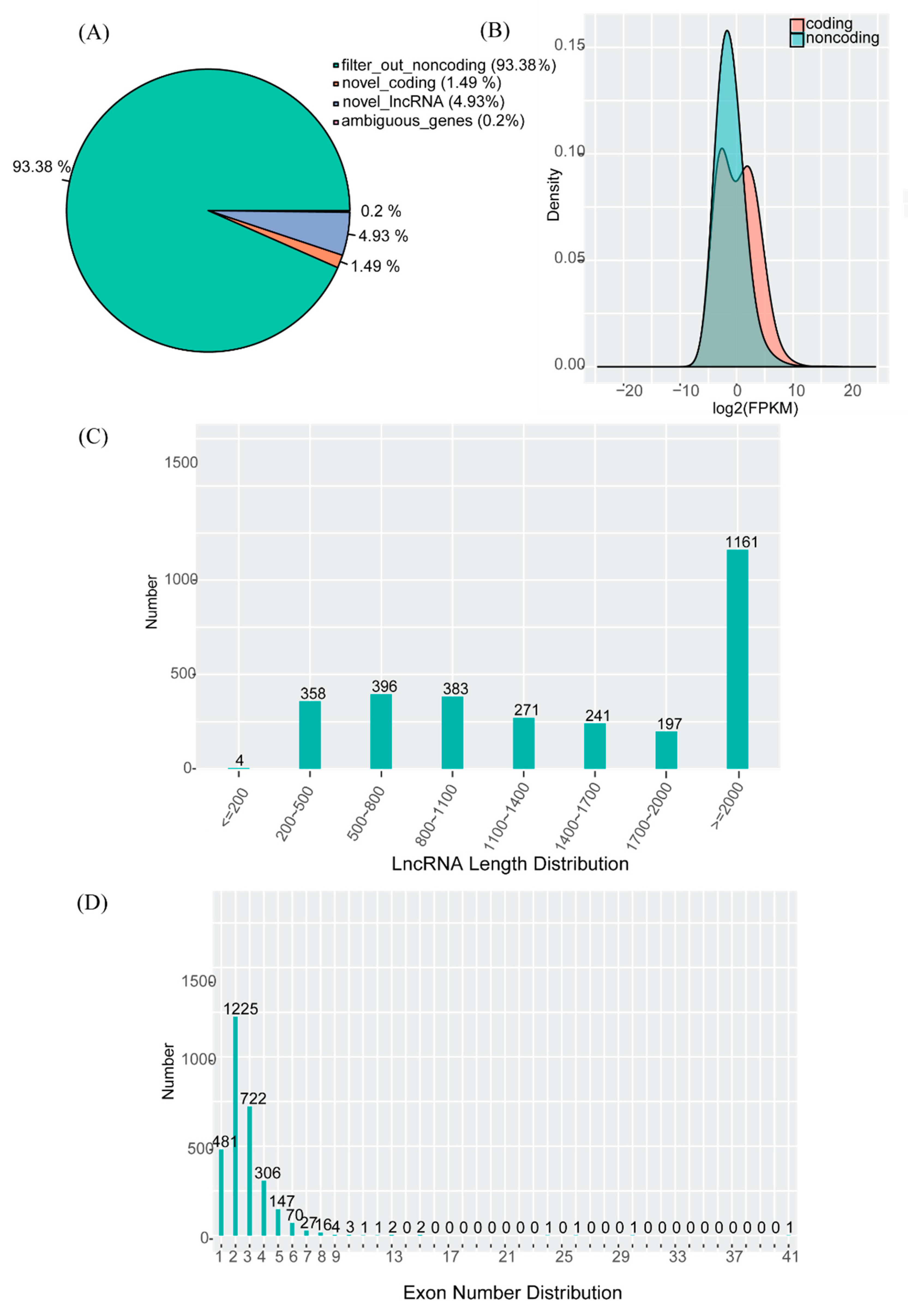
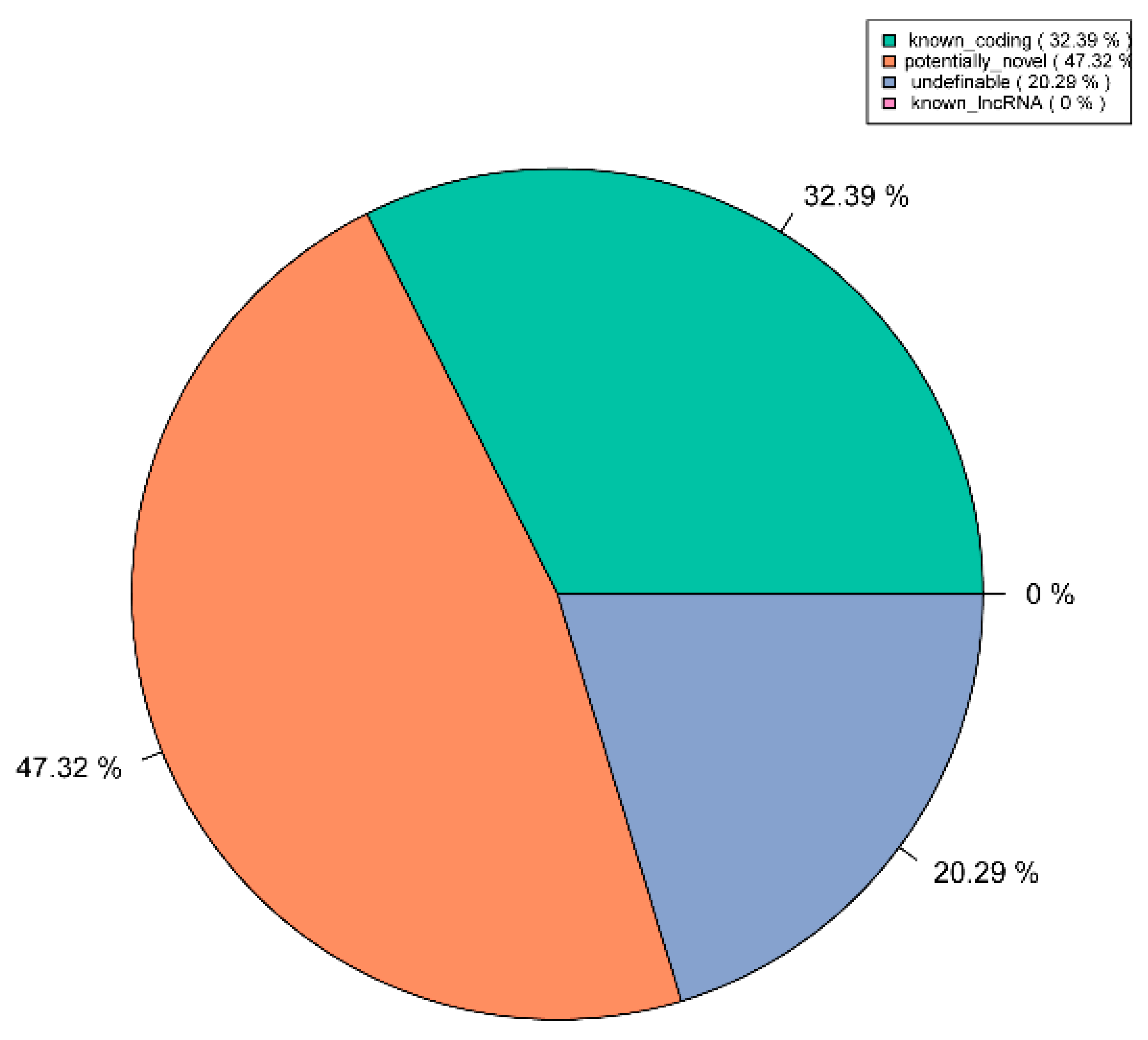
3.2. Identification of lncRNAs
3.3. Features of mRNAs and lncRNAs
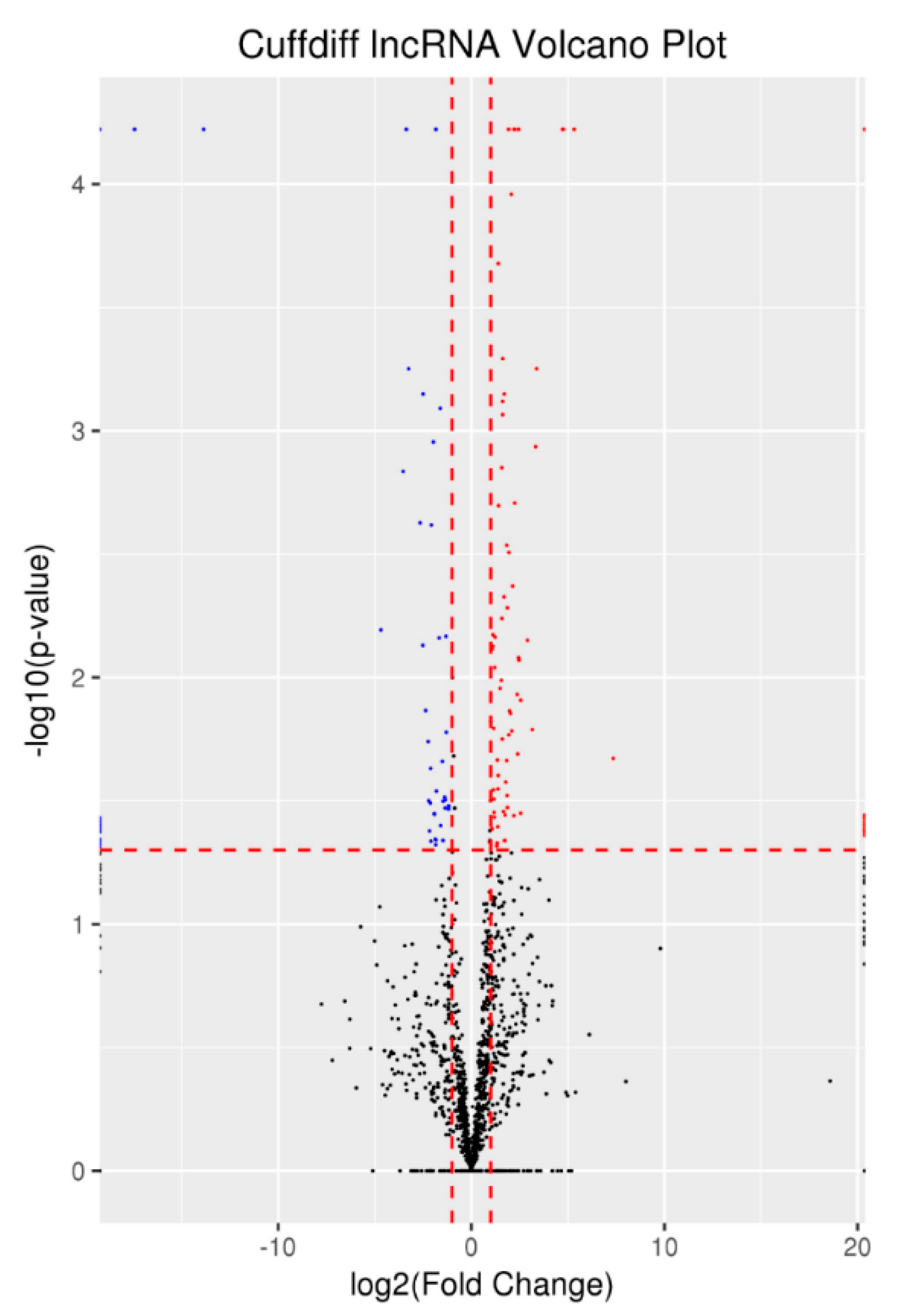
3.4. Differentially Expressed lncRNAs and mRNAs
3.5. Predication of the Targeted Genes
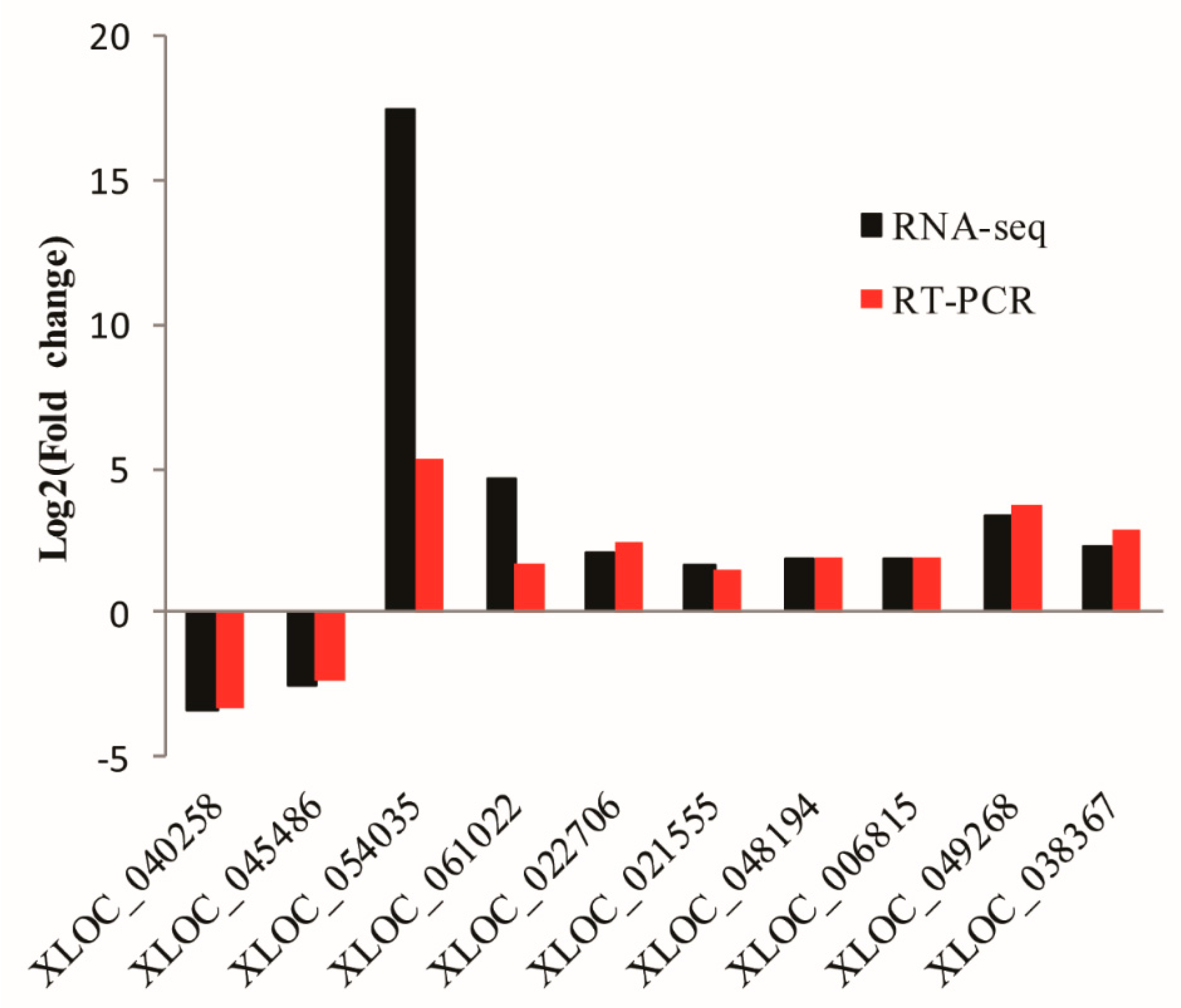
3.6. Validation of lncRNAs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
Appendix A


References
- Diamantopoulos, M.A.; Tsiakanikas, P.; Scorilas, A. Non-coding RNAs: The riddle of the transcriptome and their perspectives in cancer. Ann. Transl. Med. 2018, 6, 241. [Google Scholar] [CrossRef]
- Chen, H.; Xu, Z.; Liu, D. Small non-coding RNA and colorectal cancer. J. Cell. Mol. Med. 2019, 23, 3050–3057. [Google Scholar] [CrossRef]
- Dey, B.K.; Mueller, A.C.; Dutta, A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription 2014, 5, e944014. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.; Morton, C. Identification and function of long non-coding RNA. Front. Cell. Neurosci. 2013, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.Y.; Mansour, T.; Bellone, R.R.; Brown, C.T.; Mienaltowski, M.J.; Penedo, M.C.; Ross, P.J.; Valberg, S.J.; Murray, J.D.; Finno, C.J. Identification of long non-coding RNA in the horse transcriptome. BMC Genom. 2017, 18, 511. [Google Scholar] [CrossRef] [PubMed]
- Mansour, T.A.; Scott, E.Y.; Finno, C.J.; Bellone, R.R.; Mienaltowski, M.J.; Penedo, M.C.; Ross, P.J.; Valberg, S.J.; Murray, J.D.; Brown, C.T. Tissue resolved, gene structure refined equine transcriptome. BMC Genom. 2017, 18, 103. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, A.; Wang, L.; Li, K.; Yang, G.; He, R.; Qian, L.; Xu, N.; Huang, R.; Peng, Z.; et al. Animal genetic resources in China: Pigs. In China National Commission of Animal Genetic Resources; China Agriculture Press: Beijing, China, 2011. [Google Scholar]
- Liu, L.L.; Fang, C.; Liu, W.J. Identification on novel locus of dairy traits of Kazakh horse in Xinjiang. Gene 2018, 677, 105–110. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. 2007, 113, 1–13. [Google Scholar] [CrossRef]
- Alwasel, S.H.; Abotalib, Z.; Aljarallah, J.S.; Osmond, C.; Al Omar, S.Y.; Harrath, A.; Thornburg, K.; Barker, D.J.P. The breadth of the placental surface but not the length is associated with body size at birth. Placenta 2012, 33, 619–622. [Google Scholar] [CrossRef]
- Murphy, V.E.; Smith, R.; Giles, W.B.; Clifton, V.L. Endocrine regulation of human fetal growth: The role of the mother, placenta, and fetus. Endocr. Rev. 2006, 27, 141–169. [Google Scholar] [CrossRef] [PubMed]
- Kader, A.; Liu, X.; Dong, K.; Song, S.; Pan, J.; Yang, M.; Chen, X.; He, X.; Jiang, L.; Ma, Y. Identification of copy number variations in three Chinese horse breeds using 70K single nucleotide polymorphism BeadChip array. Anim. Genet. 2016, 47, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Ghosh, S.; Chan, C.K.K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. In Plant Bioinformatics: Methods and Protocols; Edwards, D., Ed.; Springer: New York, NY, USA, 2016; pp. 339–361. [Google Scholar]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S. Analysing RNA-Seq data with the DESeq package. Available online: https://master.bioconductor.org/help/course-materials/2011/CSAMA/Thursday/Afternoon%20Labs/DESeq.pdf (accessed on 11 January 2020).
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef]
- Zhang, G.Y.; Duan, A.G.; Zhang, J.G.; He, C.Y. Genome-wide analysis of long non-coding RNAs at the mature stage of sea buckthorn (Hippophae rhamnoides Linn) fruit. Gene 2017, 596, 130–136. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Chitwood, J.L.; Meyers-Brown, G.A.; Roser, J.F.; Ross, P.J. RNA-seq transcriptome profiling of equine inner cell mass and trophectoderm. Biol. Reprod. 2014, 90, 61. [Google Scholar] [CrossRef]
- Bamshad, M.; Le, T.; Watkins, W.S.; Dixon, M.E.; Kramer, B.E.; Roeder, A.D.; Carey, J.C.; Root, S.; Schinzel, A.; Van Maldergem, L.; et al. The spectrum of mutations in TBX3: Genotype/phenotype relationship in ulnar-mammary syndrome. Am. J. Hum. Genet. 1999, 64, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; Van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef]
- Piórkowska, K.; Żukowski, K.; Nowak, J.; Połtowicz, K.; Ropka-Molik, K.; Gurgul, A. Genome-wide RNA-Seq analysis of breast muscles of two broiler chicken groups differing in shear force. Anim. Genet. 2016, 47, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, S.S.; Park, W.C.; Ghosh, M.; Kim, J.N.; Sharma, N.; Shin, K.Y.; Cho, I.C.; Ryu, Y.C.; Oh, S.J.; Kim, S.H.; et al. Comparative transcriptomic analysis to identify differentially expressed genes in fat tissue of adult Berkshire and Jeju native pig using RNA-seq. Mol. Biol. Rep. 2014, 41, 6305–6315. [Google Scholar] [CrossRef]
- Suárez-Vega, A.; Gutiérrez-Gil, B.; Klopp, C.; Robert-Granie, C.; Tosser-Klopp, G.; Arranz, J.J. Characterization and comparative analysis of the milk transcriptome in two dairy sheep breeds using RNA sequencing. Sci. Rep. 2015, 5, 18399. [Google Scholar] [CrossRef]
- Li, J.; Weinberg, M.S.; Zerbini, L.; Prince, S.; Bronner, M. The oncogenic TBX3 is a downstream target and mediator of the TGF-β1 signaling pathway. Mol. Biol. Cell 2013, 24, 3569–3576. [Google Scholar] [CrossRef]
- Jerome-Majewska, L.A.; Achkar, T.; Luo, L.; Lupu, F.; Lacy, E. The trafficking protein Tmed2/p24β1 is required for morphogenesis of the mouse embryo and placenta. Dev. Biol. 2010, 341, 154–166. [Google Scholar] [CrossRef]
- Giudice, J.; Loehr, J.A.; Rodney, G.G.; Cooper, T.A. Alternative splicing of four trafficking genes regulates myofiber structure and skeletal muscle physiology. Cell Rep. 2016, 17, 1923–1933. [Google Scholar] [CrossRef]
- Badou, A.; Jha, M.K.; Matza, D.; Mehal, W.Z.; Freichel, M.; Flockerzi, V.; Flavell, R.A. Critical role for the β regulatory subunits of Cav channels in T lymphocyte function. Proc. Natl. Acad. Sci. USA 2006, 103, 15529–15534. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Pan, J.F.; Zhao, Q.J.; He, X.H.; Pu, Y.B.; Han, J.L.; Ma, Y.H.; Jiang, L. Detecting selection signatures on the X chromosome of the Chinese Debao pony. J. Anim. Breed. Genet. 2018, 135, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Jäderkvist Fegraeus, K.; Velie Brandon, D.; Axelsson, J.; Ang, R.; Hamilton Natasha, A.; Andersson, L.; Meadows Jennifer, R.S.; Lindgren, G. A potential regulatory region near the EDN3 gene may control both harness racing performance and coat color variation in horses. Physiol. Rep. 2018, 6, e13700. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.M.; Kim, J.W.; Kim, C.H.; An, J.H.; Johnson, A.; Song, P.I.; Rhee, S.; Choi, K.H. KAT5-mediated SOX4 acetylation orchestrates chromatin remodeling during myoblast differentiation. Cell Death Dis. 2015, 6, e1857. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.W.; Roh, K.H.; Bhandari, D.R.; Park, S.B.; Lee, S.K.; Kang, K.S. ZNF281 knockdown induced osteogenic differentiation of human multipotent stem cells in vivo and in vitro. Cell Transplant. 2013, 22, 29–40. [Google Scholar] [CrossRef]
- Makvandi-Nejad, S.; Hoffman, G.E.; Allen, J.J.; Chu, E.; Gu, E.; Chandler, A.M.; Loredo, A.I.; Bellone, R.R.; Mezey, J.G.; Brooks, S.A. Four loci explain 83% of size variation in the horse. PLoS ONE 2012, 7, e39929. [Google Scholar] [CrossRef]
- Metzger, J.; Rau, J.; Naccache, F.; Bas Conn, L.; Lindgren, G.; Distl, O. Genome data uncover four synergistic key regulators for extremely small body size in horses. BMC Genom. 2018, 19, 492. [Google Scholar] [CrossRef]
- Metzger, J.; Gast, A.C.; Schrimpf, R.; Rau, J.; Eikelberg, D.; Beineke, A.; Hellige, M.; Distl, O. Whole-genome sequencing reveals a potential causal mutation for dwarfism in the Miniature Shetland pony. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2017, 28, 143–151. [Google Scholar] [CrossRef]
- Eberth, J.E.; Graves, K.T.; MacLeod, J.N.; Bailey, E. Multiple alleles of ACAN associated with chondrodysplastic dwarfism in Miniature horses. Anim. Genet. 2018, 49, 413–420. [Google Scholar] [CrossRef]
- Frischknecht, M.; Jagannathan, V.; Plattet, P.; Neuditschko, M.; Signer-Hasler, H.; Bachmann, I.; Pacholewska, A.; Drögemüller, C.; Dietschi, E.; Flury, C.; et al. A non-synonymous HMGA2 variant decreases height in Shetland ponies and other small horses. PLoS ONE 2015, 10, e0140749. [Google Scholar] [CrossRef]
- Metzger, J.; Schrimpf, R.; Philipp, U.; Distl, O. Expression levels of LCORL are associated with body size in horses. PLoS ONE 2013, 8, e56497. [Google Scholar] [CrossRef] [PubMed]
- Norton, E.M.; Avila, F.; Schultz, N.E.; Mickelson, J.R.; Geor, R.J.; McCue, M.E. Evaluation of an HMGA2 variant for pleiotropic effects on height and metabolic traits in ponies. J. Vet. Intern. Med. 2019, 33, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Tetens, J.; Widmann, P.; Kühn, C.; Thaller, G. A genome-wide association study indicates LCORL/NCAPG as a candidate locus for withers height in German Warmblood horses. Anim. Genet. 2013, 44, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Bartholazzi Junior, A.; Quirino, C.R.; Vega, W.H.O.; Rua, M.A.S.; David, C.M.G.; Jardim, J.G. Polymorphisms in the LASP1 gene allow selection for smaller stature in ponies. Livest. Sci. 2018, 216, 160–164. [Google Scholar] [CrossRef]
- Srikanth, K.; Kim, N.Y.; Park, W.; Kim, J.M.; Kim, K.D.; Lee, K.T.; Son, J.H.; Chai, H.H.; Choi, J.W.; Jang, G.W.; et al. Comprehensive genome and transcriptome analyses reveal genetic relationship, selection signature, and transcriptome landscape of small-sized Korean native Jeju horse. Sci. Rep. 2019, 9, 16672. [Google Scholar] [CrossRef]
- Leegwater, P.A.; Vos-Loohuis, M.; Ducro, B.J.; Boegheim, I.J.; van Steenbeek, F.G.; Nijman, I.J.; Monroe, G.R.; Bastiaansen, J.W.M.; Dibbits, B.W.; van de Goor, L.H.; et al. Dwarfism with joint laxity in Friesian horses is associated with a splice site mutation in B4GALT7. BMC Genom. 2016, 17, 839. [Google Scholar] [CrossRef]
- Orr, N.; Back, W.; Gu, J.; Leegwater, P.; Govindarajan, P.; Conroy, J.; Ducro, B.; Van Arendonk, J.A.M.; MacHugh, D.E.; Ennis, S.; et al. Genome-wide SNP association–based localization of a dwarfism gene in Friesian dwarf horses. Anim. Genet. 2010, 41, 2–7. [Google Scholar] [CrossRef]
- Kader, A.; Li, Y.; Dong, K.; Irwin, D.M.; Zhao, Q.; He, X.; Liu, J.; Pu, Y.; Gorkhali, N.A.; Liu, X.; et al. Population variation reveals independent selection toward small body size in Chinese Debao pony. Genome Biol. Evol. 2016, 8, 42–50. [Google Scholar] [CrossRef]
- Emechebe, U.; Rozenberg, J.M.; Moore, B.; Firment, A.; Mirshahi, T.; Moon, A.M. T-box3 is a ciliary protein and regulates stability of the Gli3 transcription factor to control digit number. eLife 2016, 5, e07897. [Google Scholar] [CrossRef]
- Sundstrom, E.; Imsland, F.; Mikko, S.; Wade, C.; Sigurdsson, S.; Pielberg, G.R.; Golovko, A.; Curik, I.; Seltenhammer, M.H.; Solkner, J.; et al. Copy number expansion of the STX17 duplication in melanoma tissue from Grey horses. BMC Genom. 2012, 13, 365. [Google Scholar] [CrossRef]
- Snippert, H.J.; Haegebarth, A.; Kasper, M.; Jaks, V.; van Es, J.H.; Barker, N.; van de Wetering, M.; van den Born, M.; Begthel, H.; Vries, R.G.; et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 2010, 327, 1385. [Google Scholar] [CrossRef] [PubMed]
- Ansar, M.; Chung, H.-l.; Al-Otaibi, A.; Elagabani, M.N.; Ravenscroft, T.A.; Paracha, S.A.; Scholz, R.; Abdel Magid, T.; Sarwar, M.T.; Shah, S.F.; et al. Bi-allelic variants in IQSEC1 cause intellectual disability, developmental delay, and short stature. Am. J. Hum. Genet. 2019, 105, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Pousada, G.; Baloira, A.; Vilariño, C.; Cifrian, J.M.; Valverde, D. Novel mutations in BMPR2, ACVRL1 and KCNA5 genes and hemodynamic parameters in patients with pulmonary arterial hypertension. PLoS ONE 2014, 9, e100261. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, D.; Kotoh, J.; Watadani, R.; Matsumoto, K. New animal models reveal that coenzyme Q2 (Coq2) and placenta-specific 8 (Plac8) are candidate genes for the onset of type 2 diabetes associated with obesity in rats. Mamm. Genome 2015, 26, 619–629. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| lncRNA Name | Length (bp) | Primers | |
|---|---|---|---|
| XLOC_022706 | 204 | F | TTACGAGCATCGCTGGGTTT |
| R | GGTCTTACAGCGACACCACA | ||
| XLOC_021555 | 280 | F | GTTCAATGGAGGAGGGCGAA |
| R | TGGAGCATCTCGCAAAGTGT | ||
| XLOC_048194 | 215 | F | AATGCACTTTTCCGGACCCT |
| R | CCCCAGTTTGATCGTGTGGA | ||
| XLOC_006815 | 217 | F | AACGTCGGTCTCTTTTCCCC |
| R | AGCAAGTTGTTTCGGGTGGA | ||
| XLOC_049268 | 296 | F | TGTCGCCCTTCATCTCTGTG |
| R | TCAGCGGTGGAAGAAAGCAA | ||
| XLOC_038367 | 238 | F | TTGCTGGCTGTTGCTTGAAC |
| R | TCTCCATTGCCAGTTCGGTG | ||
| XLOC_040258 | 178 | F | AAACTAACCAGCTCCCCGTG |
| R | TTGCCAGCCAAAATGCCTTC | ||
| XLOC_045486 | 133 | F | CGGAACCTGTTACACTGCCT |
| R | CTCACACTTCTGCCCCACAT | ||
| XLOC_054035 | 156 | F | CGGACAATTACGCAGCCATG |
| R | CCTCTGCCCTCCTTAGTCCT | ||
| XLOC_061022 | 137 | F | CCGTCTTCCTGTTCTGCACT |
| R | ACATACACCCTGGCCTGTTG | ||
| β-ACTN (Beta-actin) | 182 | F | CAGCCTTCCTTCTTGGGTAT |
| R | TGGCATAGAGGTCTTTACGG | ||
| lncRNAs | Log2FC | p | 10 kb Upstream | 10 kb Downstream | 100 kb Upstream | 100 kb Downstream |
|---|---|---|---|---|---|---|
| XLOC_025357 | 1.84 | 5 × 10−5 | ERICH4, BCKDHA, TMEM91, DMAC2, B3GNT8, EXOSC5, B9D2, TGFB1 | |||
| XLOC_038367 | −2.24 | 5 × 10−5 | IQSEC1 | |||
| XLOC_048194 | 1.82 | 5 × 10−5 | EDN3 | |||
| XLOC_051797 | 1.61 | 8 × 10−4 | STX17 | |||
| XLOC_032225 | −3.33 | 1.15 × 10−3 | PHRF1 | LMNTD2, RASSF7 | IRF7, CDHR5, SCT, DEAF1, PHRF1, DRD4 | LMNTD2, HRAS, LOC155356, RASSF7, LRRC56, LOC121596 |
| XLOC_000736 | 3.53 | 1.45 × 10−3 | PLAU, C1H1orf55, CAMK2G | VCL | ||
| XLOC_050107 | 2.64 | 2.35 × 10−3 | IFNE | |||
| XLOC_055900 | −2.14 | 4.25 × 10−3 | IL17REL | IL17REL, TTLL8 | ALG12, PIM3, LOC11177138, CRELD2, ZBED4 | |
| XLOC_021555 | 1.66 | 6.9 × 10−3 | TBX3 | |||
| XLOC_043830 | −2.91 | 7.05 × 10−3 | MFSD1 | MFSD1, LOC15252 | ||
| XLOC_017294 | −2.38 | 0.01 | KIAA1551 | AMN1, ETFBKMT | ||
| XLOC_015361 | −2.56 | 0.01 | TEX44, NMUR1, LOC1214915 | PTMA, PDE6D | ||
| XLOC_056998 | 2.36 | 0.01 | ELF3, RNPEP, TIMM17A | GPR37L1, LOC1146338, LGR6, ARL8A, PTPN7 | ||
| XLOC_016995 | 2.23 | 0.01 | KCNA5 | |||
| XLOC_021660 | −7.36 | 0.02 | RILPL1 | TMED2 | SNRNP35, KMT5A, RILPL1, RILPL2 | TMED2, DDX55, GTF2H3, TCTN2, ATP6VA2, EIF2B1 |
| XLOC_042377 | 2.12 | 0.03 | TSN, NIFK | |||
| XLOC_059454 | 1.91 | 0.03 | CD99, XG | ZBED1, DHRSX | ||
| XLOC_008338 | −2.20 | 0.03 | COQ2, HPSE | LOC163244, PLAC8 | ||
| XLOC_022706 | 2.09 | 0.04 | TBX3 | |||
| XLOC_027025 | −1.31 | 0.04 | LOC17567 | IGSF23 | LOC17567, CEACAM19, CEACAM16 | ZNF18, IGSF23 |
| Terms | Counts | p | Gene |
|---|---|---|---|
| GO:1903445, protein transport from ciliary membrane to plasma membrane | 2 | 0.018 | RILPL1, RILPL2 |
| GO:0030335, positive regulation of cell migration | 5 | 0.022 | HRAS, FOXF1, LGR6, PLAU, TGFB1 |
| GO:2000630, positive regulation of miRNA metabolic process | 2 | 0.028 | HRAS, RELA |
| GO:0002513, tolerance induction to self-antigen | 2 | 0.028 | FOXP3, TGFB1 |
| GO:0043406, positive regulation of MAP kinase activity | 3 | 0.034 | EDN3, HRAS, TGFB1 |
| Developmental protein | 17 | 0.011 | DEAF1, USP7, CCM2, RNF17, ELF3, TBX3, CAMK2G, EN1, ACKR3, RTL1, CITED2, CXCL17, LBH, HAND1, HES5, FOXC2, OLFM1 |
| Activator | 13 | 0.015 | ATF7IP, ELF3, RELA, ZXDB, FOXP3, MED13L, KAT5, ATF7IP2, CITED2, HAND1, IRF7, FOXF1, FOXC2 |
| Transcription regulation | 31 | 0.025 | DEAF1, ZFP64, ZNF18, ELF3, ASCC1, ZXDB, ZKSCAN5, CITED2, LBH, HAND1, FOXF1, OVOL1, ZNF394, BRD9, KMT5A, ATF7IP, ZNF281, FOXL1, TBX3, RELA, GTF2H3, FOXP3, MED13L, KAT5, ATF7IP2, ZNF789, HES5, DMRTC2, IRF7, FOXC2, NR5A2 |
| ecb05205:Proteoglycans in cancer | 6 | 0.029 | HRAS, HPSE, CAMK2G, SDC4, PLAU, TGFB1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, Y.; Zhang, Y.; Zhang, T.; Han, J.; Ma, Y.; Liu, X. Identification of Novel lncRNAs Differentially Expressed in Placentas of Chinese Ningqiang Pony and Yili Horse Breeds. Animals 2020, 10, 119. https://doi.org/10.3390/ani10010119
Pu Y, Zhang Y, Zhang T, Han J, Ma Y, Liu X. Identification of Novel lncRNAs Differentially Expressed in Placentas of Chinese Ningqiang Pony and Yili Horse Breeds. Animals. 2020; 10(1):119. https://doi.org/10.3390/ani10010119
Chicago/Turabian StylePu, Yabin, Yanli Zhang, Tian Zhang, Jianlin Han, Yuehui Ma, and Xuexue Liu. 2020. "Identification of Novel lncRNAs Differentially Expressed in Placentas of Chinese Ningqiang Pony and Yili Horse Breeds" Animals 10, no. 1: 119. https://doi.org/10.3390/ani10010119
APA StylePu, Y., Zhang, Y., Zhang, T., Han, J., Ma, Y., & Liu, X. (2020). Identification of Novel lncRNAs Differentially Expressed in Placentas of Chinese Ningqiang Pony and Yili Horse Breeds. Animals, 10(1), 119. https://doi.org/10.3390/ani10010119