
Artificial Rearing of Atlantic Salmon Juveniles for Supportive Breeding Programs Induces Long-Term Effects on Gut Microbiota after Stocking

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. Sequences and Statistical Analyses
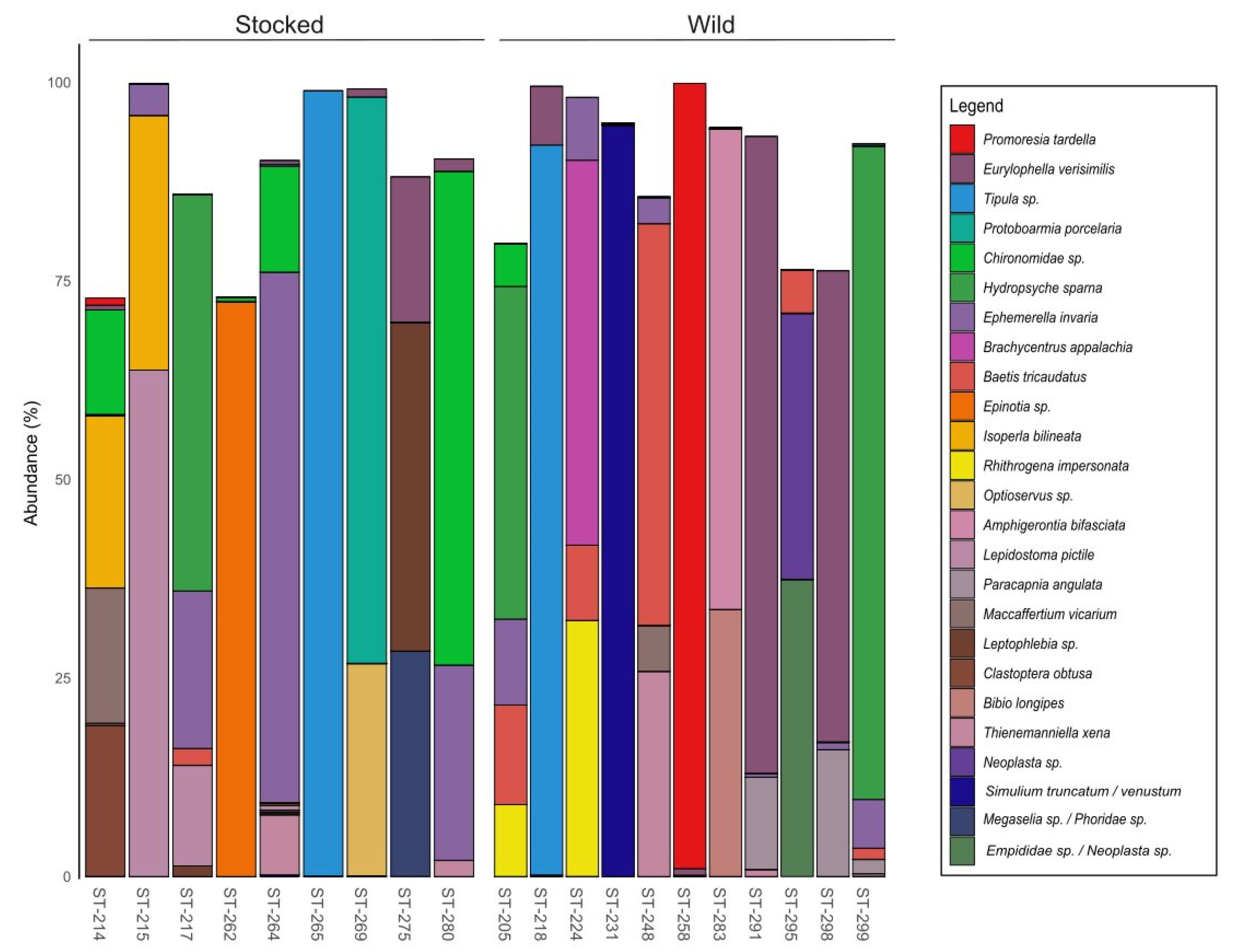
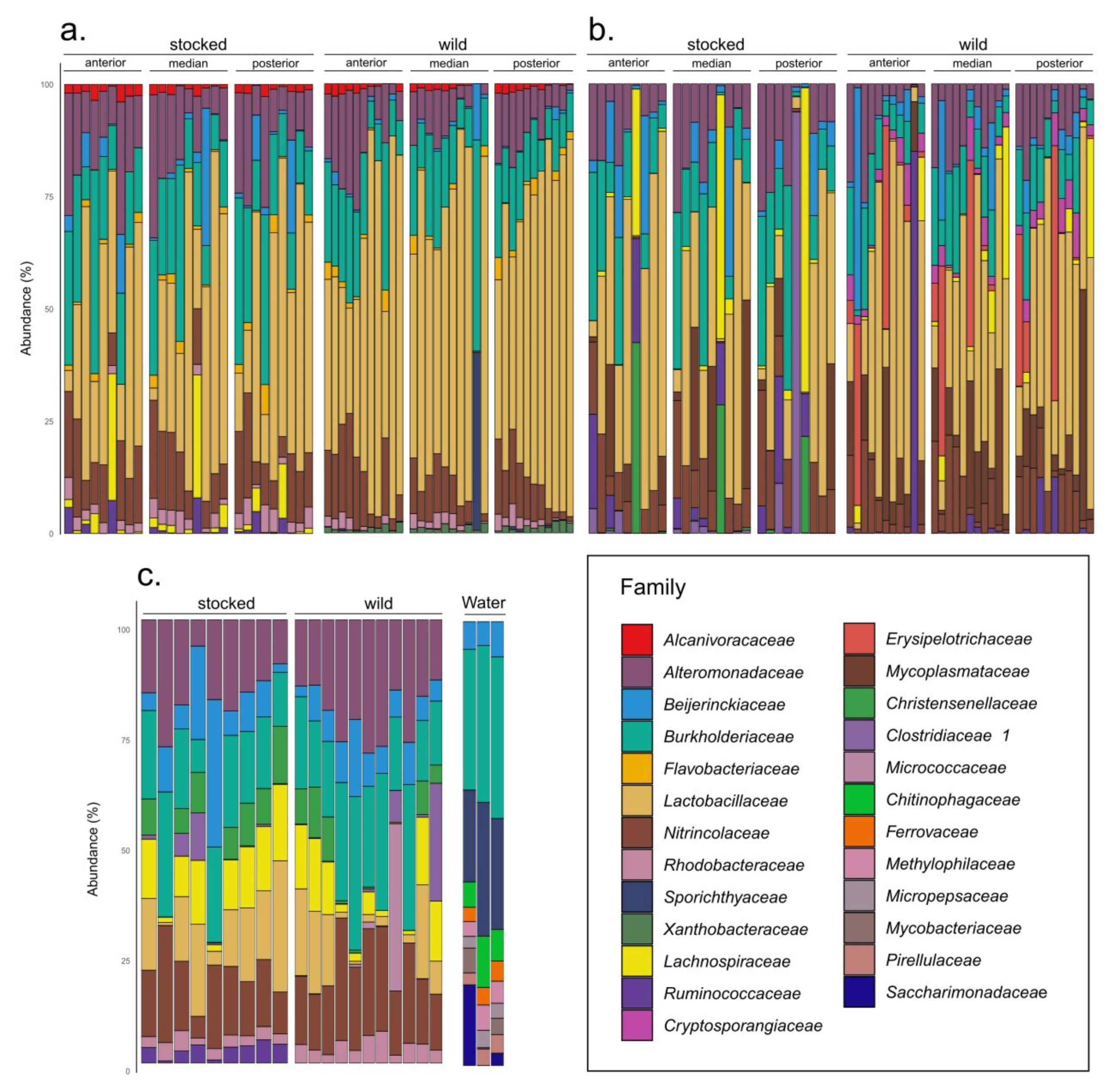
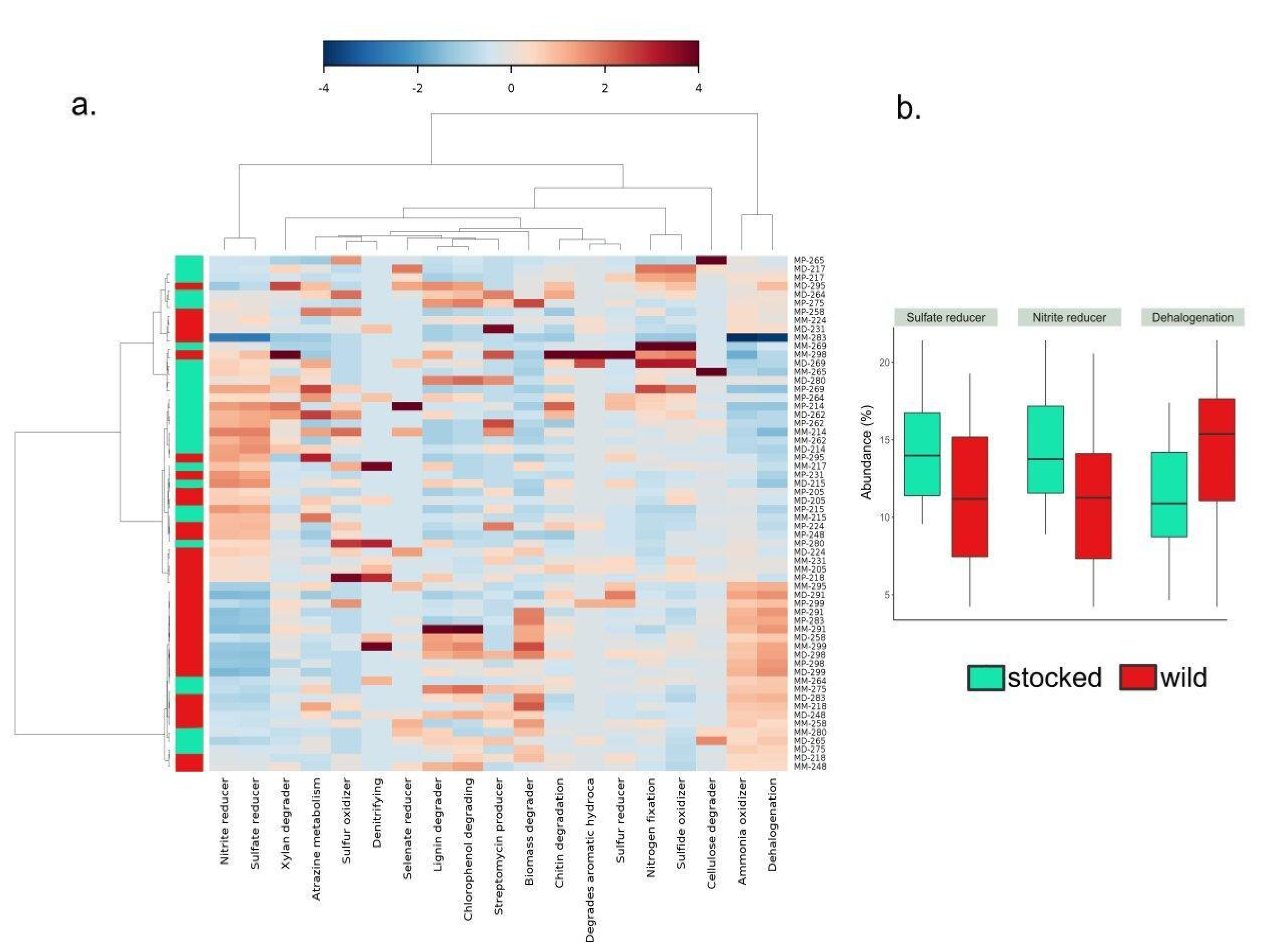
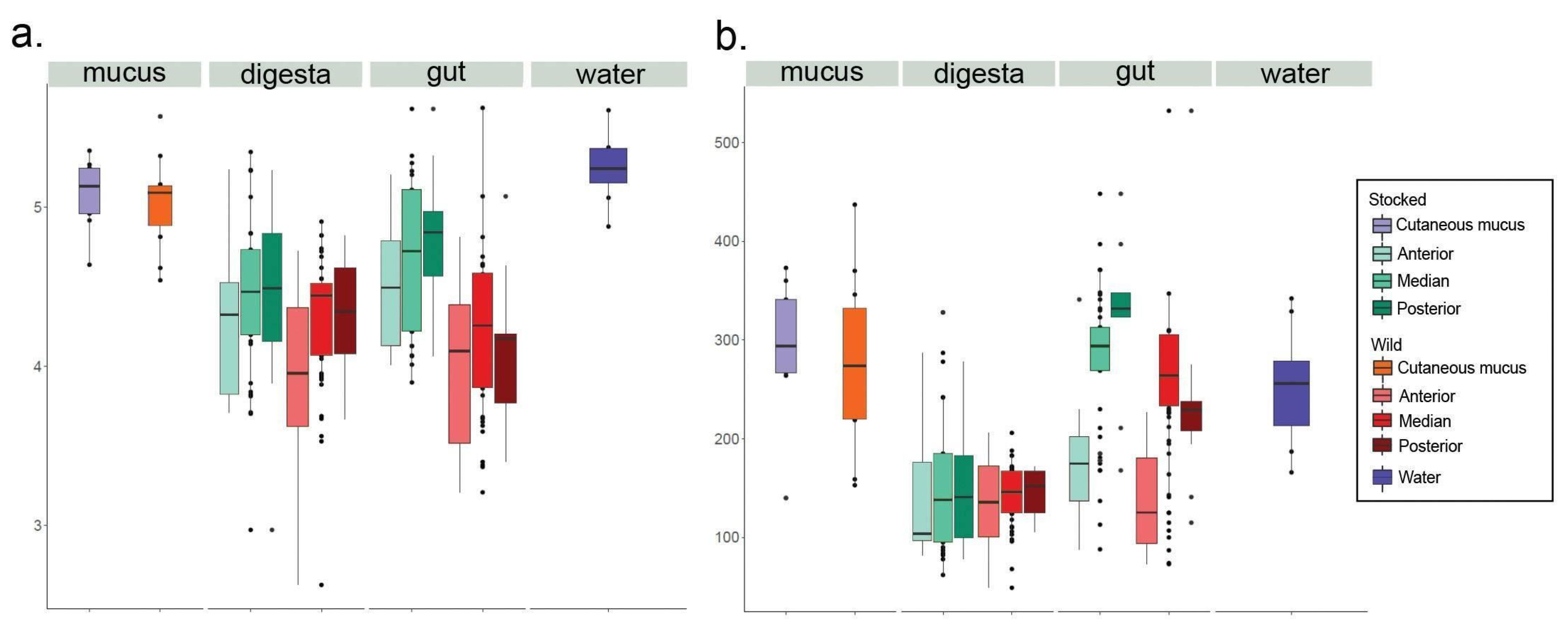
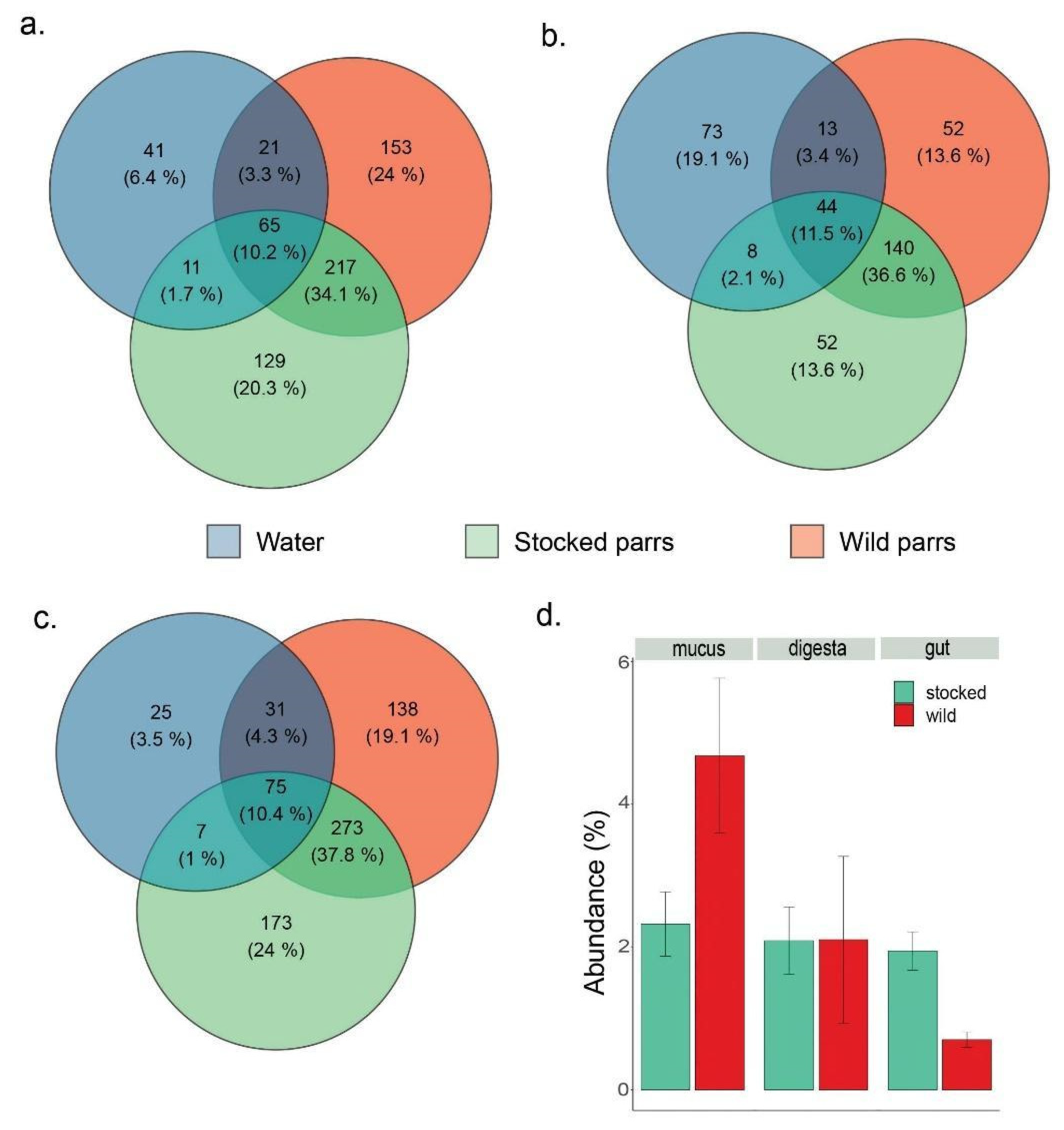
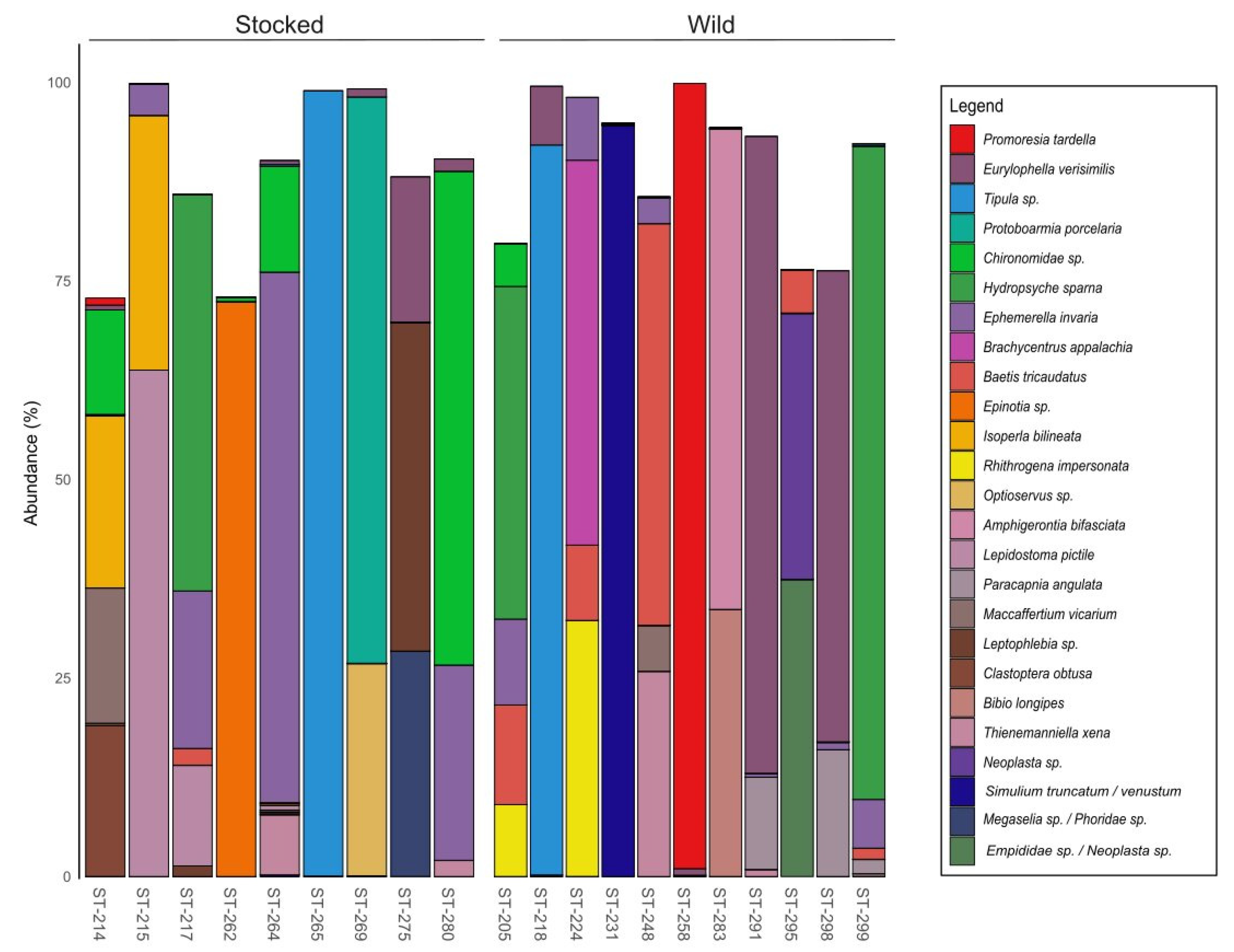
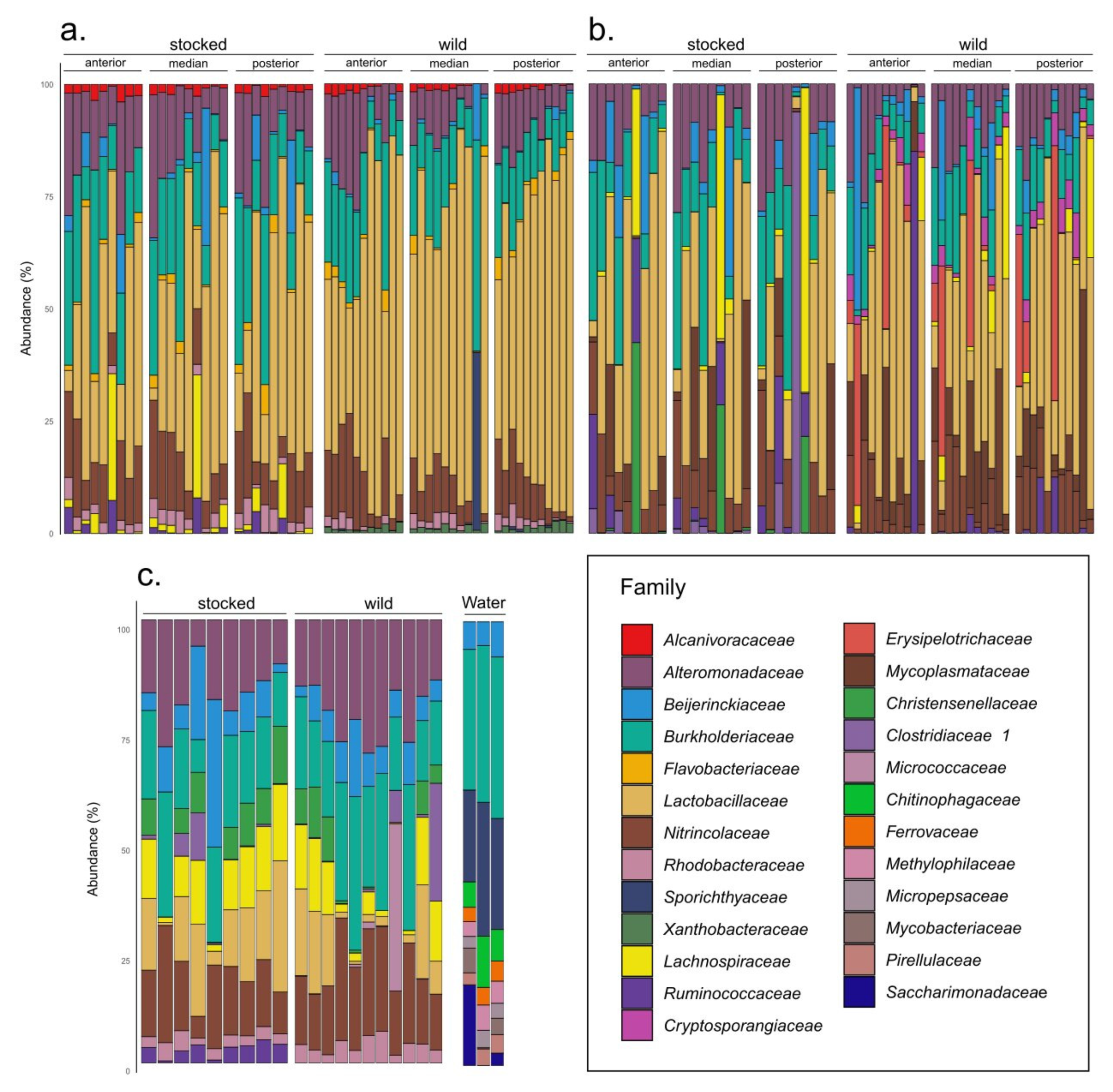
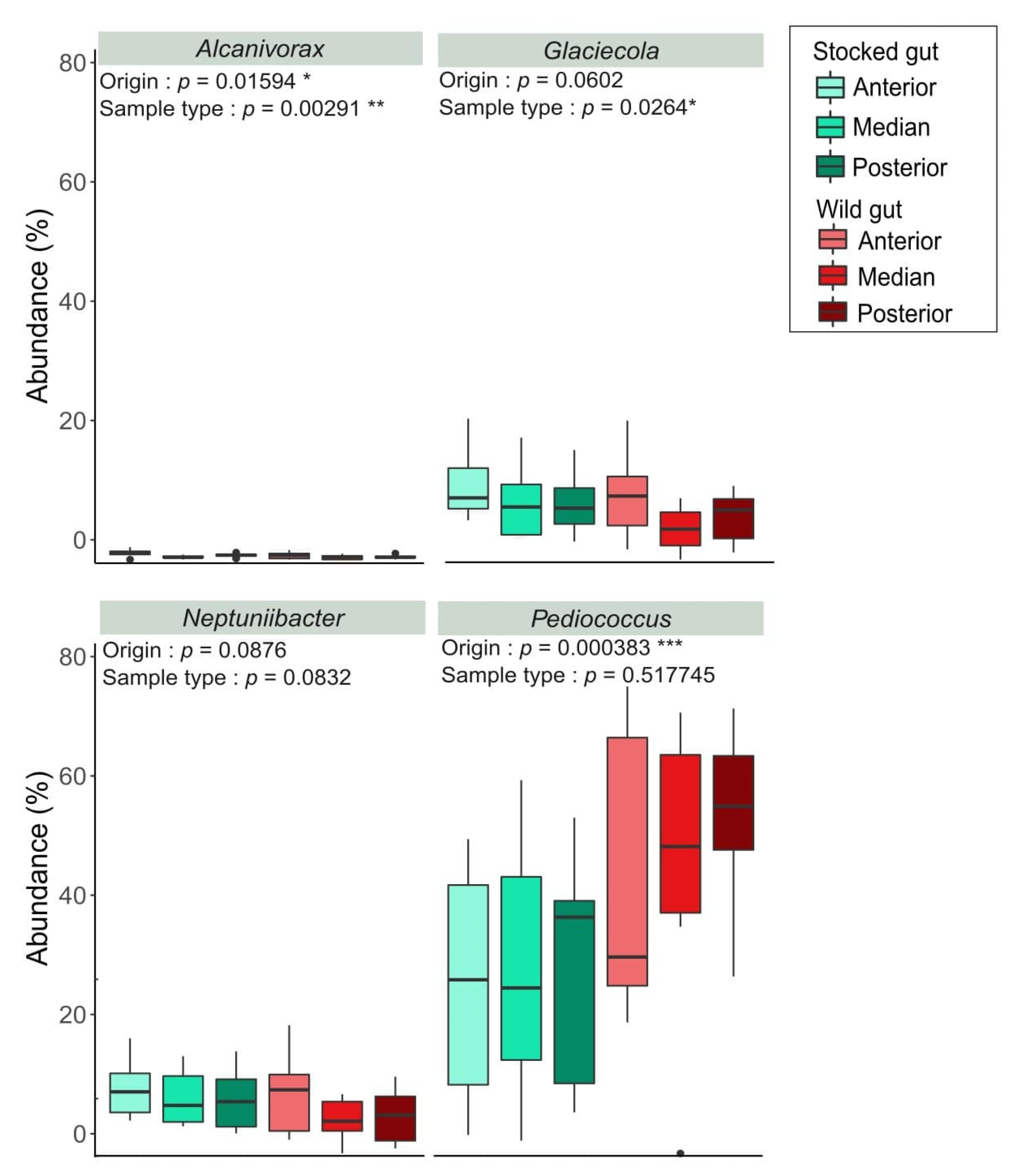
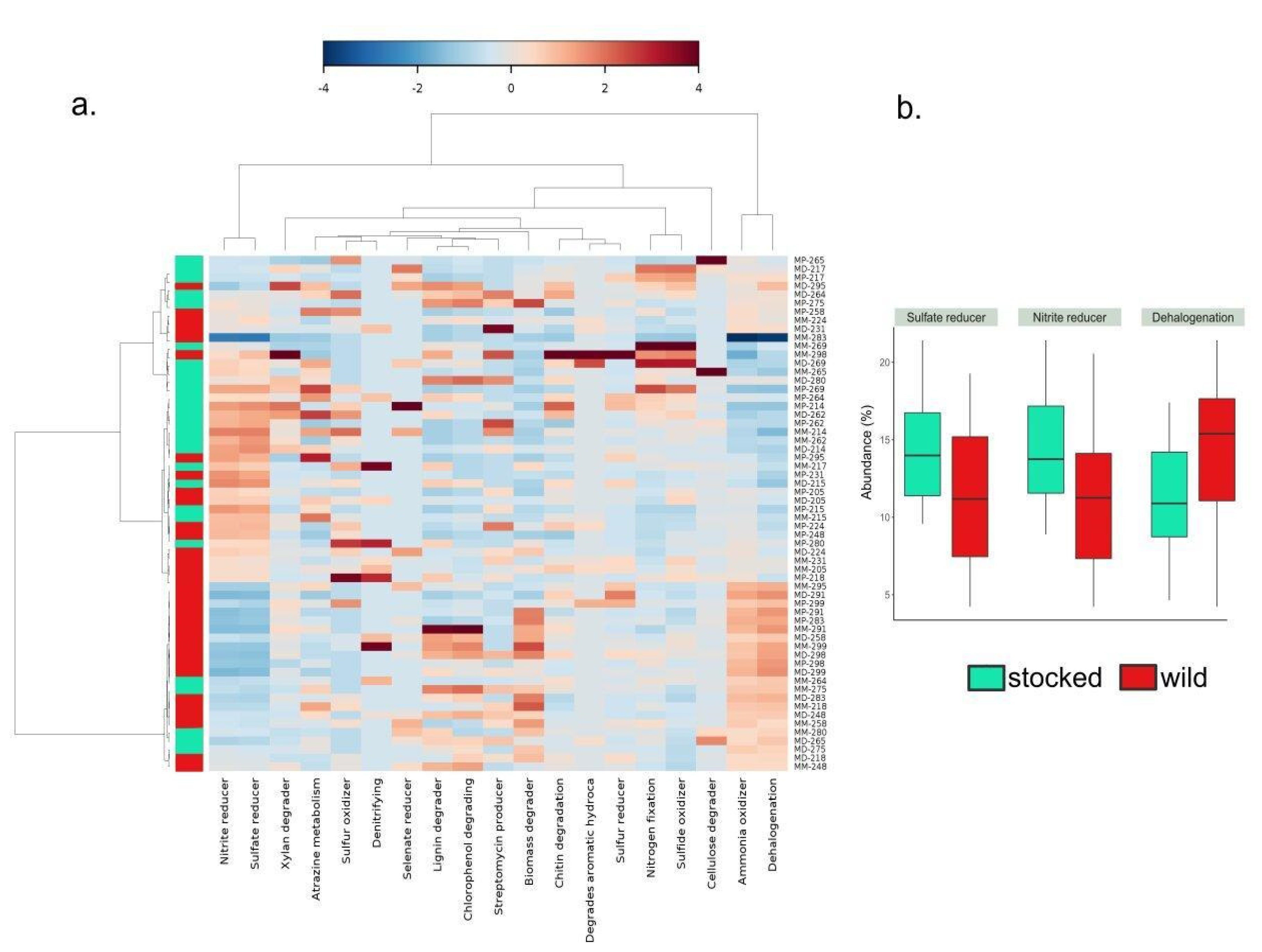
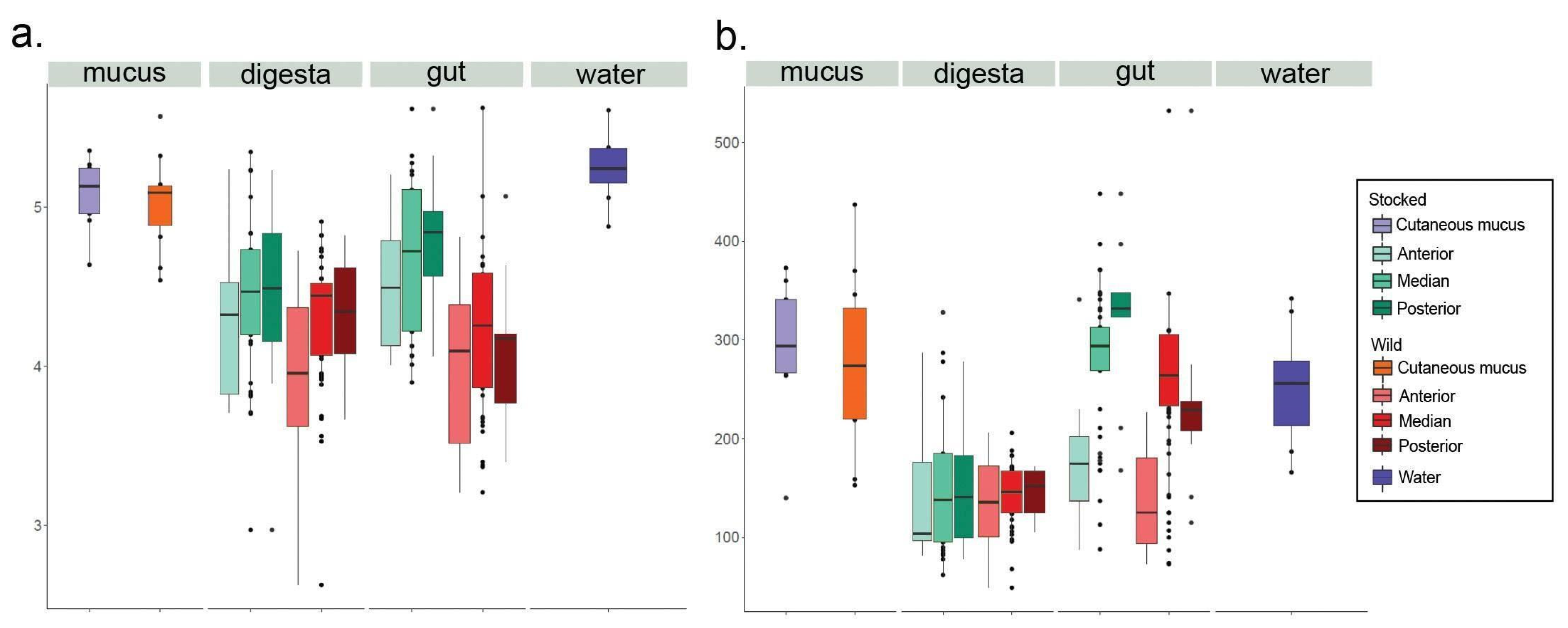
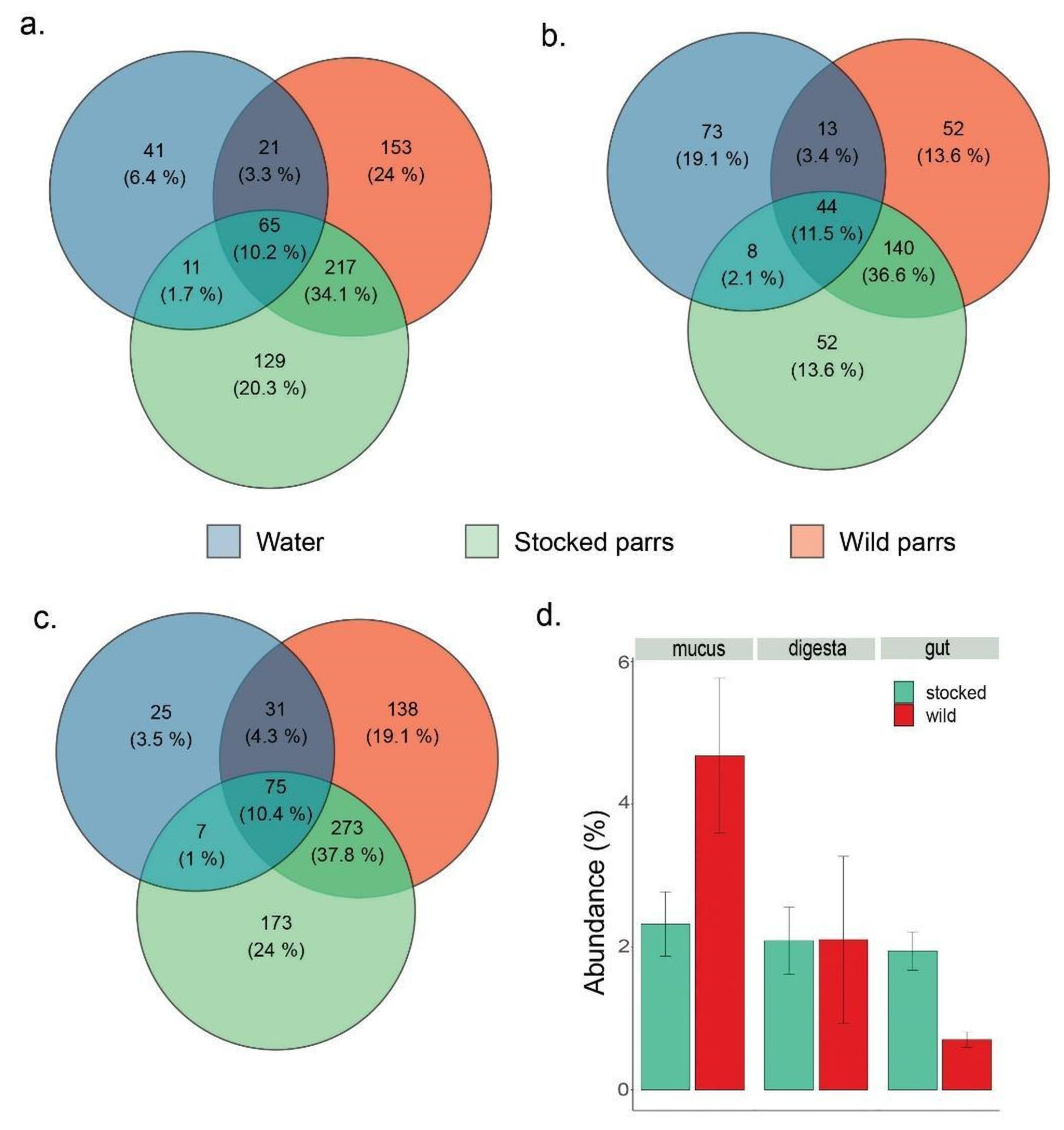
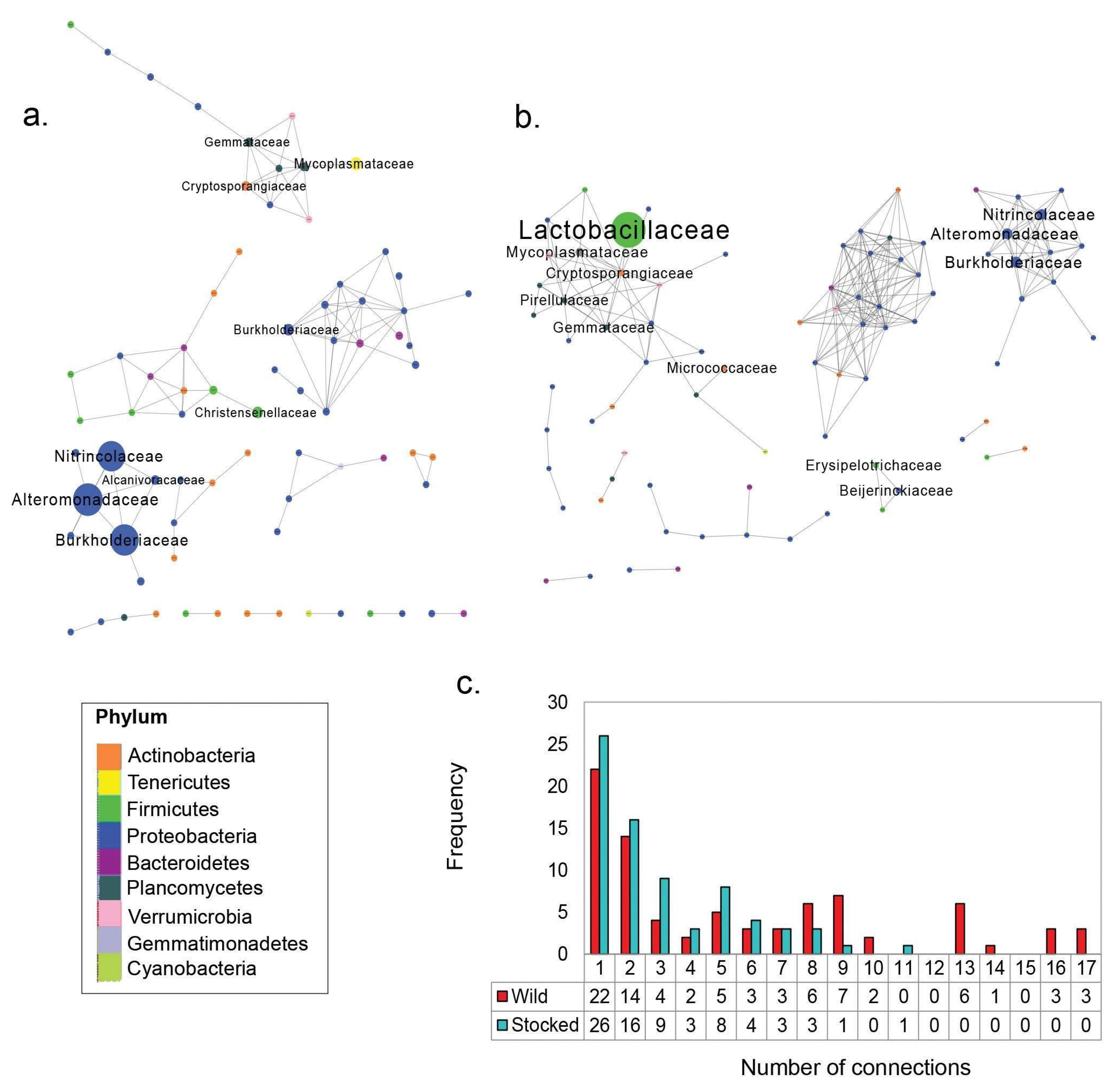
3. Results
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alenghat, T. Epigenomics and the Microbiota. Toxicol. Pathol. 2015, 43, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Cortese, R.; Lu, L.; Yu, Y.; Ruden, D.; Claud, E.C. Epigenome-Microbiome Crosstalk: A Potential New Paradigm Influencing Neonatal Susceptibility to Disease. Epigenetics 2016, 11, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Spena, J.R.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-Term Health Development. Front. Pediatrics 2017, 5, 178. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.L.; Zhong, H.Z.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; Lieber, A.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, Birth Mode, and Diet Shape Microbiome Maturation during Early Life. Sci. Transl. Med. 2016, 8, 343ra82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, R.I.; Sghir, A.; Gaskins, H.R. Developmental Microbial Ecology of the Neonatal Gastrointestinal Tract. Am. J. Clin. Nutr. 1999, 69, 1035s–1045s. [Google Scholar] [CrossRef]
- Nagpal, R.; Tsuji, H.; Takahashi, T.; Nomoto, K.; Kawashima, K.; Nagata, S.; Yamashiro, Y. Ontogenesis of the Gut Microbiota Composition in Healthy, Full-Term, Vaginally Born and Breast-Fed Infants over the First 3 Years of Life: A Quantitative Bird’s-Eye View. Front. Microbiol. 2017, 8, 1388. [Google Scholar] [CrossRef] [Green Version]
- Sylvain, F.-É.; Derome, N. Vertically and Horizontally Transmitted Microbial Symbionts Shape the Gut Microbiota Ontogenesis of a Skin-Mucus Feeding Discus Fish Progeny. Sci. Rep. 2017, 7, 5263. [Google Scholar] [CrossRef] [Green Version]
- Burns, A.R.; Stephens, W.Z.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. Contribution of Neutral Processes to the Assembly of Gut Microbial Communities in the Zebrafish over Host Development. ISME J. 2016, 10, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Hansen, G.H.; Olafsen, J.A. Bacterial Colonization of Cod (Gadus morhua L.) and Halibut (Hippoglossus hippoglossus) Eggs in Marine Aquaculture. Appl. Environ. Microbiol. 1989, 55, 1435–1446. [Google Scholar] [CrossRef] [Green Version]
- Hansen, G.H.; Olafsen, J.A. Bacterial Interactions in Early Life Stages of Marine Cold Water Fish. Microb. Ecol. 1999, 38, 1–26. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost Microbiomes: The State of the Art in Their Characterization, Manipulation and Importance in Aquaculture and Fisheries. Front. Microbiol. 2014, 5, 207. [Google Scholar] [CrossRef] [Green Version]
- Dionne, M.; Miller, K.M.; Dodson, J.J.; Caron, F.; Bernatchez, L. Clinal Variation in Mhc Diversity with Temperature: Evidence for the Role of Host–Pathogen Interaction on Local Adaptation in Atlantic Salmon. Evolution 2007, 61, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Rawls, J.F.; Mahowald, M.A.; Ley, R.E.; Gordon, J.I. Reciprocal Gut Microbiota Transplants from Zebrafish and Mice to Germ-Free Recipients Reveal Host Habitat Selection. Cell 2006, 127, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Boutin, S.; Sauvage, C.; Bernatchez, L.; Audet, C.; Derome, N. Inter Individual Variations of the Fish Skin Microbiota: Host Genetics Basis of Mutualism? edited by S. Mariani. PLoS ONE 2014, 9, e102649. [Google Scholar] [CrossRef] [PubMed]
- Uren Webster, T.M.; Consuegra, S.; Hitchings, M.; Garcia de Leaniz, C. Interpopulation Variation in the Atlantic Salmon Microbiome Reflects Environmental and Genetic Diversity. Appl. Environ. Microbiol. 2018, 84, e00691-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoetendal, E.G.; Antoon, D.L.; Akkermans, W.M.; Akkermans-van, V.; Arjan, J.; De Visser, G.M.; De Vos, W.M. The Host Genotype Affects the Bacterial Community in the Human Gastrointestinal Tract. Microb. Ecol. Health Dis. 2001, 13, 129–134. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; McGinnity, P.; Dionne, M.; Letourneau, J.; Thonier, F.; Carvalho, G.R.; Creer, S.; Derome, N. The Biogeography of the Atlantic Salmon (Salmo salar) Gut Microbiome. ISME J. 2016, 10, 1280–1284. [Google Scholar] [CrossRef] [Green Version]
- Lokesh, J.; Kiron, V.; Sipkema, D.; Fernandes, J.M.O.; Moum, T. Succession of Embryonic and the Intestinal Bacterial Communities of Atlantic Salmon (Salmo salar) Reveals Stage-Specific Microbial Signatures. MicrobiologyOpen 2018, 8, e00672. [Google Scholar] [CrossRef]
- Dehler, C.E.; Secombes, C.J.; Martin, S.A.M. Environmental and Physiological Factors Shape the Gut Microbiota of Atlantic Salmon Parr (Salmo salar L.). Aquaculture 2017, 467, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Landeira-Dabarca, A.; Sieiro, C.; Álvarez, M. Change in Food Ingestion Induces Rapid Shifts in the Diversity of Microbiota Associated with Cutaneous Mucus of Atlantic Salmon Salmo salar: Effect of Diet on S. salar Mucous Microbiota. J. Fish. Biol. 2013, 82, 893–906. [Google Scholar] [CrossRef]
- Conlon, M.; Bird, A. The Impact of Diet and Lifestyle on Gut Microbiota and Human Health. Nutrients 2014, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.R.; Links, M.G.; Collins, S.A.; Mansfield, G.S.; Drew, M.D.; Van Kessel, A.G.; Hill, J.E. Effects of Plant-Based Diets on the Distal Gut Microbiome of Rainbow Trout (Oncorhynchus mykiss). Aquaculture 2012, 350–353, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Gajardo, K.; Rodiles, A.; Kortner, T.M.; Krogdahl, Å.; Bakke, A.M.; Merrifield, D.L.; Sørum, H. A High-Resolution Map of the Gut Microbiota in Atlantic Salmon (Salmo salar): A Basis for Comparative Gut Microbial Research. Sci. Rep. 2016, 6, 6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartviksen, M.; Vecino, J.L.G.; Ringø, E.; Bakke, A.M.; Wadsworth, S.; Krogdahl, Å.; Ruohonen, K.; Kettunen, A. Alternative Dietary Protein Sources for Atlantic Salmon (Salmo salar L.) Effect on Intestinal Microbiota, Intestinal and Liver Histology and Growth. Aquac. Nutr. 2014, 20, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Sylvain, F.-É.; Holland, A.; Bouslama, S.; Audet-Gilbert, É.; Lavoie, C.; Val, A.L.; Derome, N. Fish Skin and Gut Microbiomes Show Contrasting Signatures of Host Species and Habitat. Appl Environ Microbiol. 2020, 86, e00789-20. [Google Scholar] [CrossRef]
- Sylvain, F.-É.; Cheaib, B.; Llewellyn, M.; Correia, T.G.; Fagundes, D.B.; Val, A.L.; Derome, N. pH Drop Impacts Differentially Skin and Gut Microbiota of the Amazonian Fish Tambaqui (Colossoma macropomum). Sci. Rep. 2016, 6, 32032. [Google Scholar] [CrossRef] [Green Version]
- Vrieze, A.; Out, C.; Fuentes, S.; Jonker, L.; Reuling, I.; Kootte, R.S.; van Nood, E.; Holleman, F.; Knaapen, M.; Romijn, J.A.; et al. Impact of Oral Vancomycin on Gut Microbiota, Bile Acid Metabolism, and Insulin Sensitivity. J. Hepatol. 2014, 60, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Rawls, J.F. Intestinal Microbiota Composition in Fishes Is Influenced by Host Ecology and Environment. Mol. Ecol. 2012, 21, 3100–3102. [Google Scholar] [CrossRef] [Green Version]
- FQSA (Fédération Québécoise pour le Saumon Atlantique). Mémoire sur le Saumon Atlantique au Québec; Chambre des Communes: Ottawa, ON, Canada, 2015. [Google Scholar]
- Lavoie, C.; Courcelle, M.; Redivo, B.; Derome, N. Structural and Compositional Mismatch between Captive and Wild Atlantic Salmon (Salmo salar) Parrs’ Gut Microbiota Highlights the Relevance of Integrating Molecular Ecology for Management and Conservation Methods. Evol. Appl. 2018, 11, 1671–1685. [Google Scholar] [CrossRef]
- Dionne, M.; Caron, F.; Dodson, J.J.; Bernatchez, L. Landscape Genetics and Hierarchical Genetic Structure in Atlantic Salmon: The Interaction of Gene Flow and Local Adaptation: Landscape Genetics in Atlantic Salmon. Mol. Ecol. 2008, 17, 2382–2396. [Google Scholar] [CrossRef] [PubMed]
- Araki, H.; Berejikian, B.A.; Ford, M.J.; Blouin, M.S. SYNTHESIS: Fitness of Hatchery-Reared Salmonids in the Wild: Fitness of Hatchery Fish. Evol. Appl. 2008, 1, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Ford, M.J. Selection in Captivity during Supportive Breeding May Reduce Fitness in the Wild. Conserv. Biol. 2002, 16, 815–825. [Google Scholar] [CrossRef]
- Milot, E.; Perrier, C.; Papillon, L.; Dodson, J.J.; Bernatchez, L. Reduced Fitness of Atlantic Salmon Released in the Wild after One Generation of Captive Breeding. Evol. Appl. 2013, 6, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Stringwell, R.; Lock, A.; Stutchbury, C.J.; Baggett, E.; Taylor, J.; Gough, P.J.; Garcia de Leaniz, C. Maladaptation and Phenotypic Mismatch in Hatchery-Reared Atlantic Salmon Salmo salar Released in the Wild. J. Fish. Biol. 2014, 85, 1927–1945. [Google Scholar] [CrossRef]
- Ingerslev, H.C.; von Gersdorff Jørgensen, L.; Lenz Strube, M.; Larsen, N.; Dalsgaard, I.; Boye, M.; Madsen, L. The Development of the Gut Microbiota in Rainbow Trout (Oncorhynchus mykiss) Is Affected by First Feeding and Diet Type. Aquaculture 2014, 424–425, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Lokesh, J.; Kiron, V. Transition from Freshwater to Seawater Reshapes the Skin-Associated Microbiota of Atlantic Salmon. Sci. Rep. 2016, 6, 19707. [Google Scholar] [CrossRef] [Green Version]
- Cheaib, B.; Seghouani, H.; Ijaz, U.Z.; Derome, N. Community Recovery Dynamics in Yellow Perch Microbiome after Gradual and Constant Metallic Perturbations. Microbiome 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Q.; Li, J.; Yu, Y.; Wang, J.; He, Z.; Van Nostrand, J.D.; Kempher, M.L.; Wu, L.; Wang, Y.; Liao, L.; et al. Environmental Filtering Decreases with Fish Development for the Assembly of Gut Microbiota. Environ. Microbiol. 2016, 18, 4739–4754. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chow, K.; Fleming, E.; Oh, J. Selective Colonization Ability of Human Fecal Microbes in Different Mouse Gut Environments. ISME J. 2019, 13, 805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Lei, F.; Zhu, L.; Li, S.; Wu, Z.; Zhang, R.; Gao, G.F.; Zhu, B.; Wang, X. Exposure of Different Bacterial Inocula to Newborn Chicken Affects Gut Microbiota Development and Ileum Gene Expression. ISME J. 2010, 4, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylvain, F.-É.; Holland, A.; Audet-Gilbert, É.; Val, A.L.; Derome, N. Amazon Fish Bacterial Communities Show Structural Convergence along Widespread Hydrochemical Gradients. Mol. Ecol. 2019, 28, 3612–3626. [Google Scholar] [CrossRef]
- CCPA. Lignes Directrices sur: Le soin Et L’utilisation des Poissons en Recherche, en Enseignement et Dans les Tests; CCPA: Ottawa, ON, Canada, 2005.
- O’reilly, P.T.; Hamilton, L.C.; Mcconnell, S.K.; Wright, J.M. Rapid analysis of genetic variation in Atlantic salmon (Salmo salar) by PCR multiplexing of dinucleotide and tetranucleotide microsatellites. Can. J. Fish. Aquat. Sci. 1996, 53, 2292–2298. [Google Scholar] [CrossRef]
- King, T.L.; Eackles, M.S.; Letcher, B.H. Microsatellite DNA Markers for the Study of Atlantic Salmon (Salmo salar) Kinship, Population Structure, and Mixed-Fishery Analyses. Mol. Ecol. Notes 2005, 5, 130–132. [Google Scholar] [CrossRef]
- Slettan, A.; Olsaker, I.; Lie, Ø. Atlantic Salmon, Salmo salar, Microsatellites at the SSOSL25, SSOSL85, SSOSL311, SSOSL417 Loci. Anim. Genet. 1995, 26, 281–282. [Google Scholar] [CrossRef] [PubMed]
- Paterson, S.; Piertney, S.B.; Knox, D.; Gilbey, J.; Verspoor, E. Characterization and PCR Multiplexing of Novel Highly Variable Tetranucleotide Atlantic Salmon (Salmo salar L.) Microsatellites. Mol. Ecol. Notes 2004, 4, 160–162. [Google Scholar] [CrossRef]
- Presa, P.; Guyomard, R. Conservation of Microsatellites in Three Species of Salmonids. J. Fish. Biol. 1996, 49, 1326–1329. [Google Scholar] [CrossRef]
- Aljanabi, S.M.; Martinez, I. Universal and Rapid Salt-Extraction of High Quality Genomic DNA for PCR-Based Techniques. Nucleic Acids Res. 1997, 25, 4692–4693. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising How the Computer Program CERVUS Accommodates Genotyping Error Increases Success in Paternity Assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Goudet, J. hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Geller, J.; Meyer, C.; Parker, M.; Hawk, H. Redesign of PCR Primers for Mitochondrial Cytochrome c Oxidase Subunit I for Marine Invertebrates and Application in All-Taxa Biotic Surveys. Mol. Ecol. Resour. 2013, 13, 851–861. [Google Scholar] [CrossRef]
- Leray, M.; Yang, J.Y.; Meyer, C.P.; Mills, S.C.; Agudelo, N.; Ranwez, V.; Boehm, J.T.; Machida, R.J. A New Versatile Primer Set Targeting a Short Fragment of the Mitochondrial COI Region for Metabarcoding Metazoan Diversity: Application for Characterizing Coral Reef Fish Gut Contents. Front. Zool. 2013, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing Data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Jianguo, X.; Yifeng, L.; You, Z.; An, C.G.; Cruz, J.A.; Sinelnikov, I.; Budwill, K.; Nesbø, C.L.; Wishart, D.S. METAGENassist: A Comprehensive Web Server for Comparative Metagenomics. Nucleic Acids Res. 2012, 40, W88–W95. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P.; Van Domselaar, G.; Shrivastava, S.; Guo, A.; O’Neill, B.; Cruz, J.; Ellison, M.; Wishart, D.S. BacMap: An Interactive Picture Atlas of Annotated Bacterial Genomes. Nucleic Acids Res. 2005, 33, D317–D320. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Barrett, T.; Benson, D.A.; Bolton, E.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Federhen, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012, 40, D13–D25. [Google Scholar] [CrossRef] [Green Version]
- Pagani, I.; Liolios, K.; Jansson, I.; Chen, M.A.; Smirnova, T.; Nosrat, B.; Markowitz, V.M.; Kyrpides, N.C. The Genomes OnLine Database (GOLD) v.4: Status of Genomic and Metagenomic Projects and Their Associated Metadata. Nucleic Acids Res. 2012, 40, D571–D579. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams (2007–2015). Available online: https://bioinfogp.cnb.csic.es/tool/venny/index.html (accessed on 8 April 2019).
- Czerniawski, R.M.; Pilecka-Rapacz, J.D. Stocking experiment with Atlantic salmon and sea trout parr reared on either live prey or a pellet diet. J. Appl. Ichthyol. 2011, 27, 984–989. [Google Scholar] [CrossRef]
- Fleming, I.A.; Lamberg, A.; Jonsson, B. Effects of Early Experience on the Reproductive Performance of Atlantic Salmon. Behav. Ecol. 1997, 8, 470–480. [Google Scholar] [CrossRef]
- Jacobsen, J. Feeding Habits of Wild and Escaped Farmed Atlantic Salmon, Salmo salar L., in the Northeast Atlantic. ICES J. Mar. Sci. 2001, 58, 916–933. [Google Scholar] [CrossRef] [Green Version]
- Le Luyer, J.; Laporte, M.; Beacham, T.D.; Kaukinen, K.H.; Withler, R.E.; Leong, J.S.; Rondeau, E.B.; Koop, B.F.; Bernatchez, L. Parallel Epigenetic Modifications Induced by Hatchery Rearing in a Pacific Salmon. Proc. Natl. Acad. Sci. USA 2017, 114, 12964–12969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlov, A.; Gerasimov, Y.; Lapshin, O. The Feeding Behaviour of Cultured and Wild Atlantic Salmon, Salmo salar L., in the Louvenga River, Kola Peninsula, Russia. ICES J. Mar. Sci. 2006, 63, 1297–1303. [Google Scholar] [CrossRef]
- Piggins, D.J.; Mills, C.P.R. Comparative Aspects of the Biology of Naturally Produced and Hatchery-Reared Atlantic Salmon Smolts (Salmo salar L.). Aquaculture 1985, 45, 321–333. [Google Scholar] [CrossRef]
- Poole, W.R.; Nolan, D.T.; Wevers, T.; Dillane, M.; Cotter, D.; Tully, O. An Ecophysiological Comparison of Wild and Hatchery-Raised Atlantic Salmon (Salmo salar L.) Smolts from the Burrishoole System, Western Ireland. Aquaculture 2003, 222, 301–314. [Google Scholar] [CrossRef]
- Sevellec, M.; Laporte, M.; Bernatchez, A.; Derome, N.; Bernatchez, L. Evidence for host effect on the intestinal microbiota of whitefish (Coregonus sp.) species pairs and their hybrids. Ecol. Evol. 2019, 9, 11762–11774. [Google Scholar] [PubMed] [Green Version]
- Stephens, W.Z.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. The Composition of the Zebrafish Intestinal Microbial Community Varies across Development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponisio, L.; Fernanda, C.; Valdovinos, S.; Allhoff, K.T.; Gaiarsa, M.P.; Barner, A.; Guimarães, P.R.; Hembry, D.H.; Morrison, B.; Gillespie, R. A Network Perspective for Community Assembly. Front. Ecol. Evol. 2019, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Schryver, P.D.; Vadstein, O. Ecological Theory as a Foundation to Control Pathogenic Invasion in Aquaculture. ISME J. 2014, 8, 2360–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weighted | Unweighted | |||
|---|---|---|---|---|
| F | p-Value | F | p-Value | |
| Total microbiota | 3.0842 | 0.011 * | 1.6813 | 0.015 * |
| Digesta | 1.8822 | 0.069 | 1.8704 | 0.002 ** |
| Gut | 5.8617 | 0.003 ** | 1.2358 | 0.072 |
| Skin mucus | 1.6048 | 0.133 | 1.0326 | 0.313 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavoie, C.; Wellband, K.; Perreault, A.; Bernatchez, L.; Derome, N. Artificial Rearing of Atlantic Salmon Juveniles for Supportive Breeding Programs Induces Long-Term Effects on Gut Microbiota after Stocking. Microorganisms 2021, 9, 1932. https://doi.org/10.3390/microorganisms9091932
Lavoie C, Wellband K, Perreault A, Bernatchez L, Derome N. Artificial Rearing of Atlantic Salmon Juveniles for Supportive Breeding Programs Induces Long-Term Effects on Gut Microbiota after Stocking. Microorganisms. 2021; 9(9):1932. https://doi.org/10.3390/microorganisms9091932
Chicago/Turabian StyleLavoie, Camille, Kyle Wellband, Alysse Perreault, Louis Bernatchez, and Nicolas Derome. 2021. "Artificial Rearing of Atlantic Salmon Juveniles for Supportive Breeding Programs Induces Long-Term Effects on Gut Microbiota after Stocking" Microorganisms 9, no. 9: 1932. https://doi.org/10.3390/microorganisms9091932
APA StyleLavoie, C., Wellband, K., Perreault, A., Bernatchez, L., & Derome, N. (2021). Artificial Rearing of Atlantic Salmon Juveniles for Supportive Breeding Programs Induces Long-Term Effects on Gut Microbiota after Stocking. Microorganisms, 9(9), 1932. https://doi.org/10.3390/microorganisms9091932