
Assessment of Bacillus subtilis Plasmid pLS20 Conjugation in the Absence of Quorum Sensing Repression

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Primers
2.2. Plasmid and Strain Construction
2.3. Conjugation
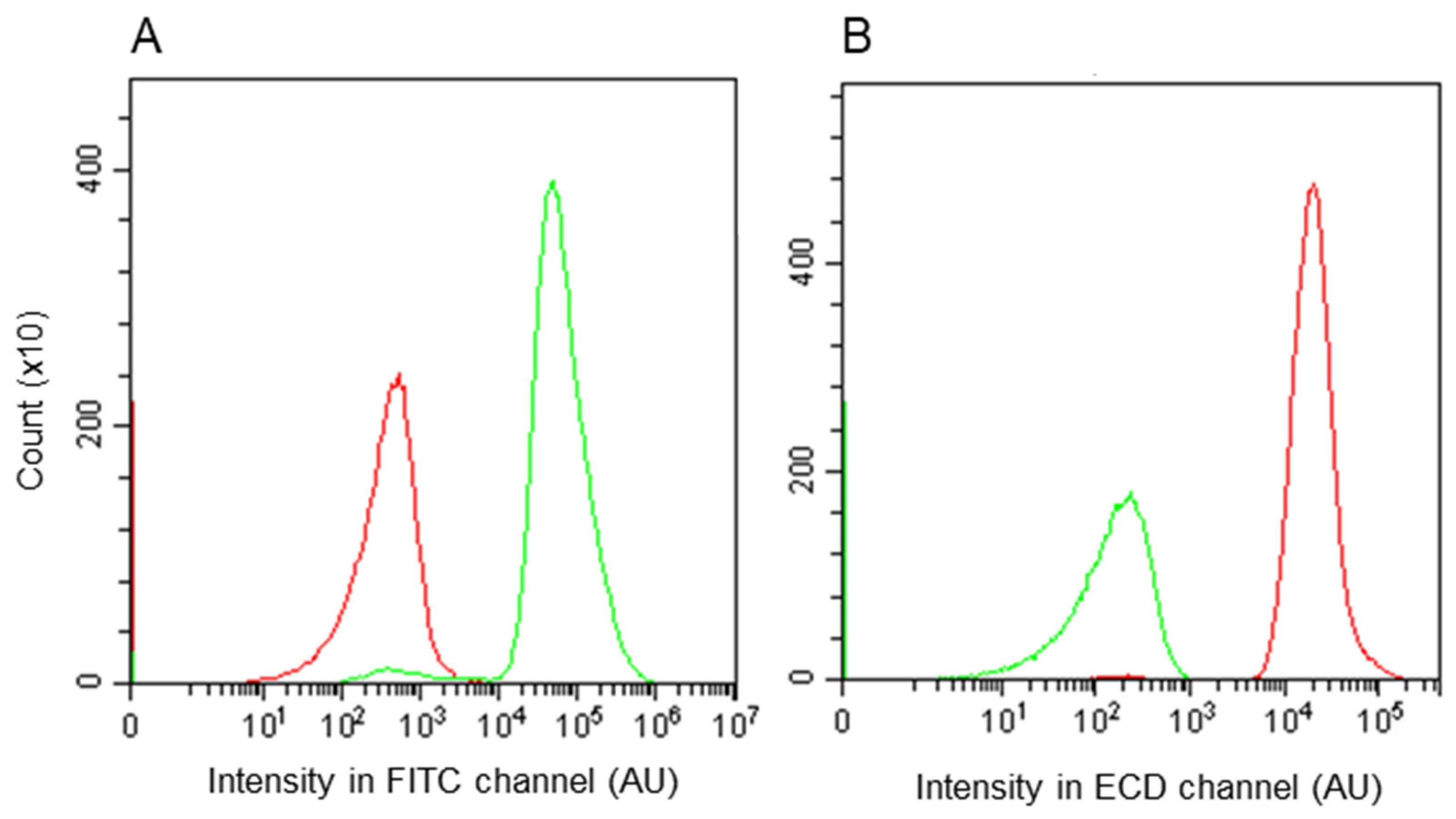
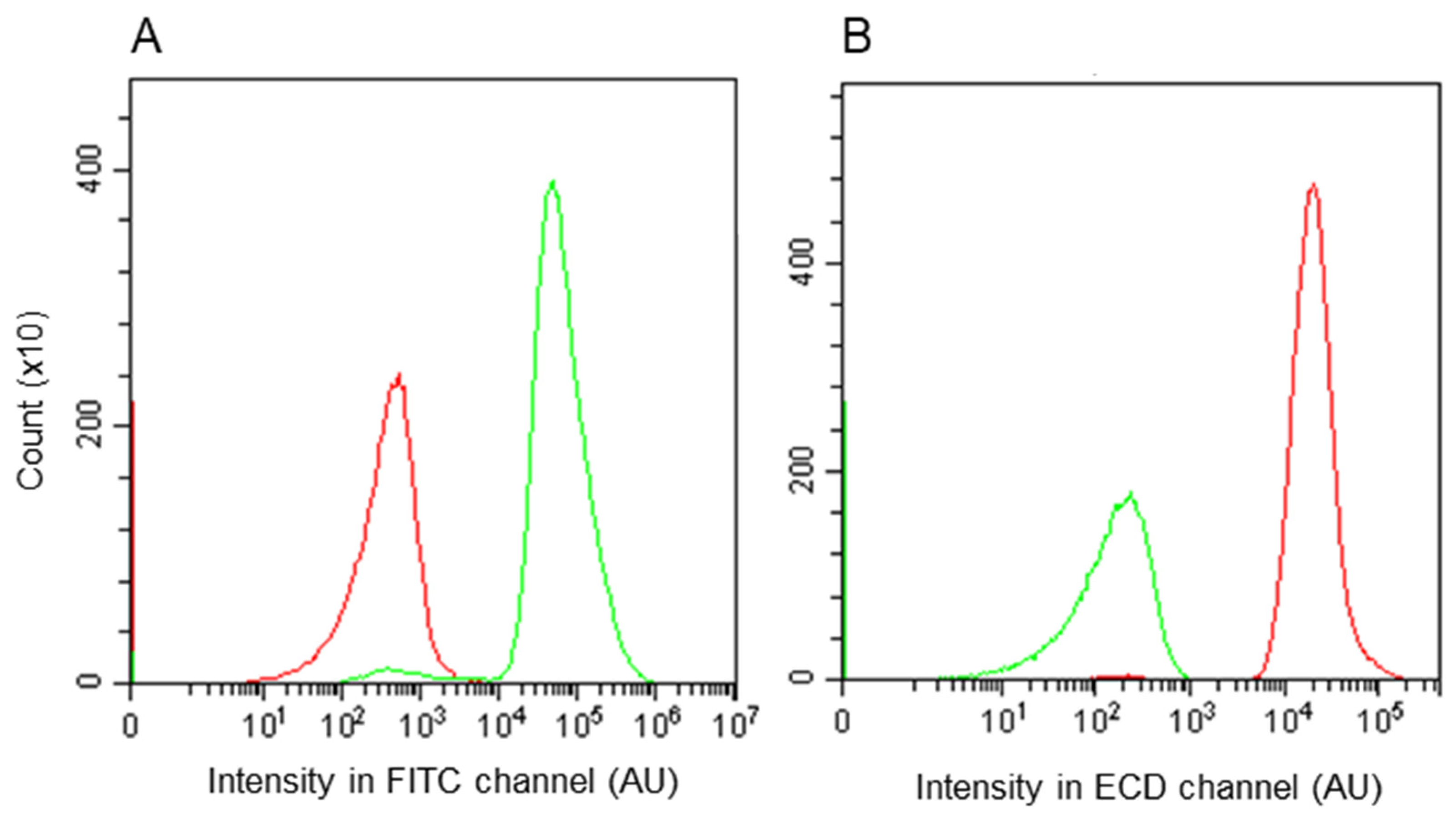
2.4. Flow Cytometric Analysis
2.5. Microscopy
3. Results
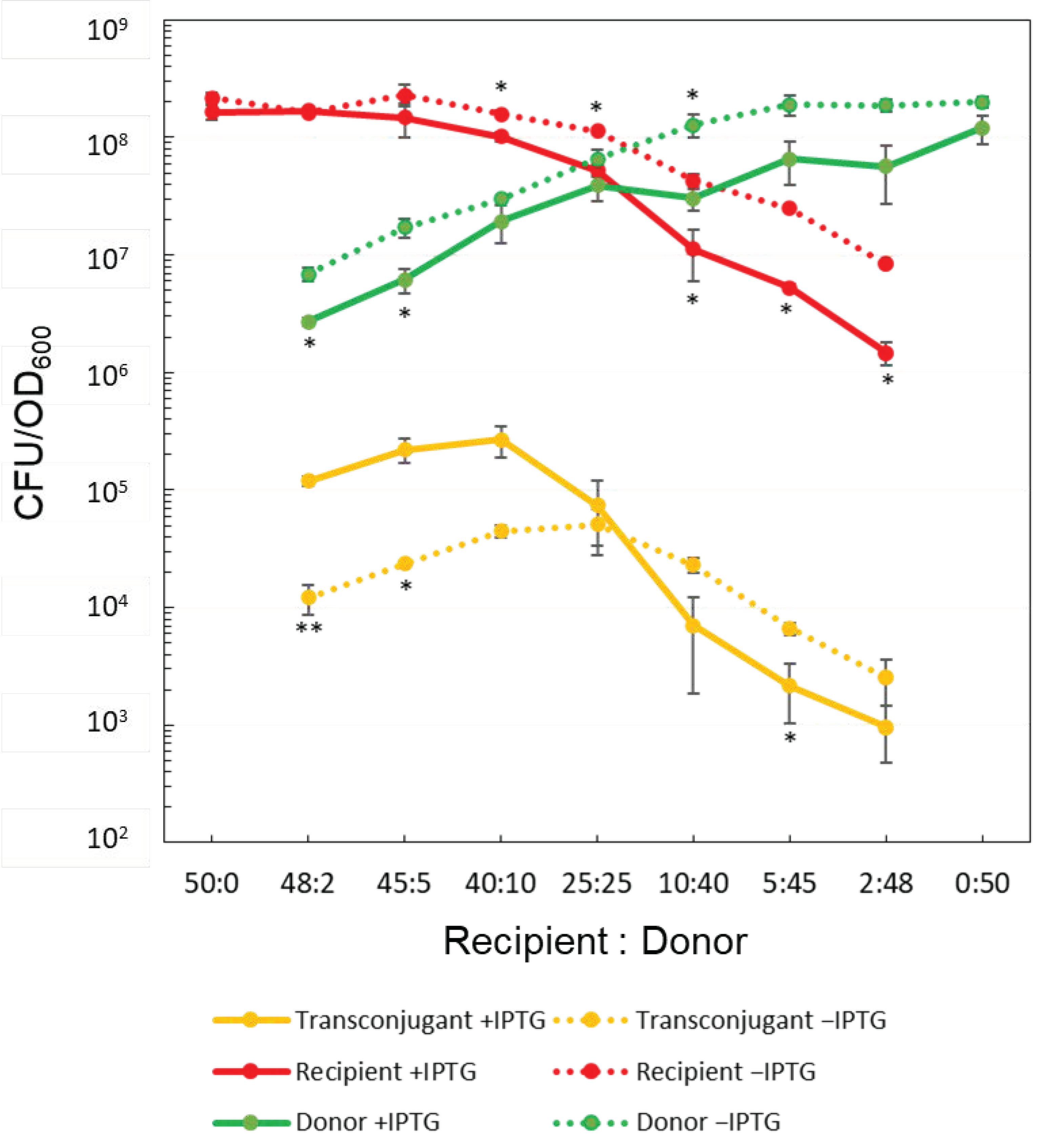
3.1. Conventional Colony Formation Assay
- (i)
- The CFU/OD600 values of both donors and recipients tended to decrease when the donors had been grown in the presence of IPTG compared to its absence.
- (ii)
- In the presence of IPTG, the CFU/OD600 of transconjugants was higher only when the concentration of recipients exceeded that of donors, whereas, in the absence of IPTG, the CFU/OD600 of transconjugants gave an apparent peak at recipients: donors = 25:25, and the value similar to that were found in the presence of IPTG.
- (iii)
- As the number of donors exceeded that of recipients, the CFU/OD600 of transconjugants showed a tendency to be lower in the presence of IPTG than in its absence.
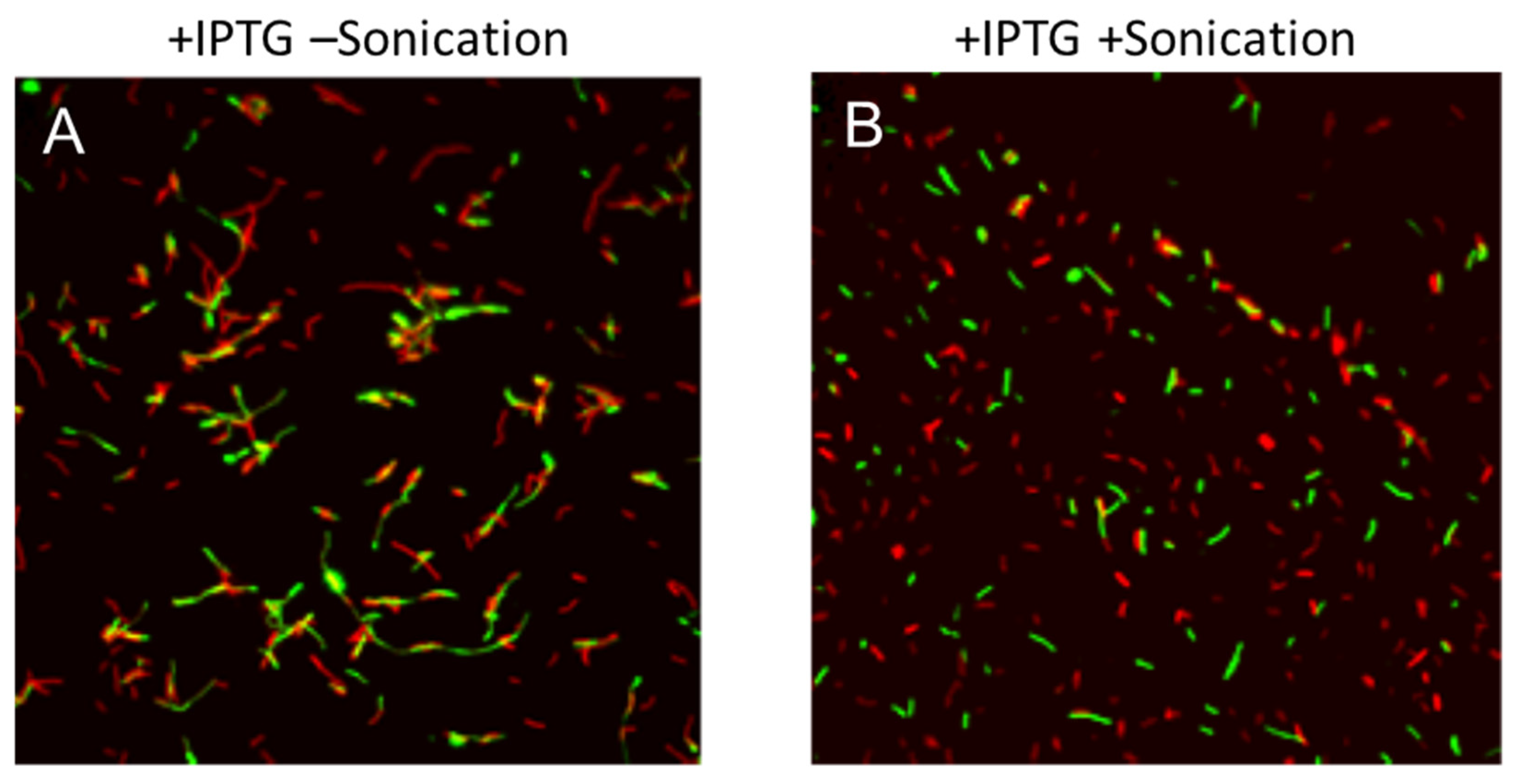
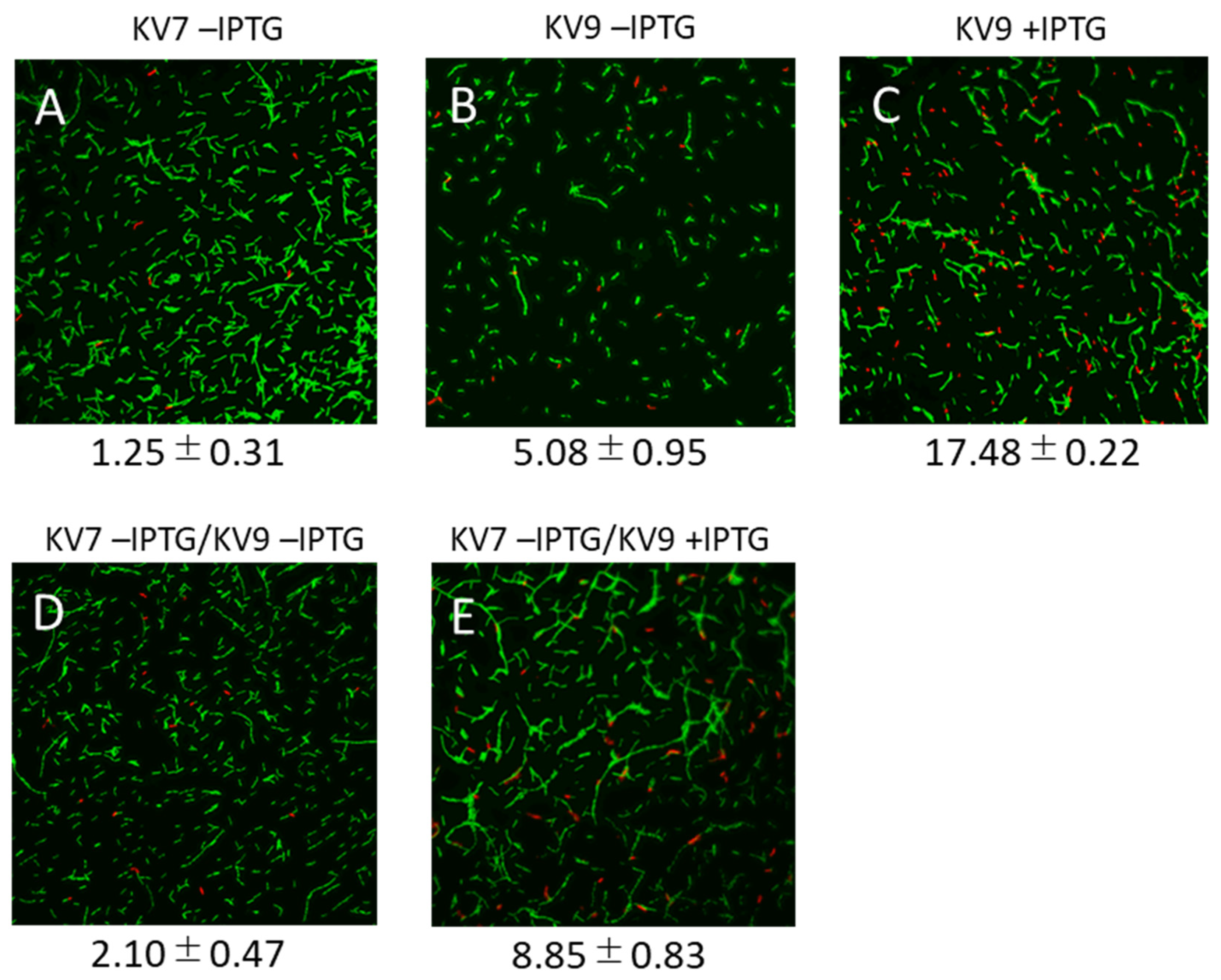
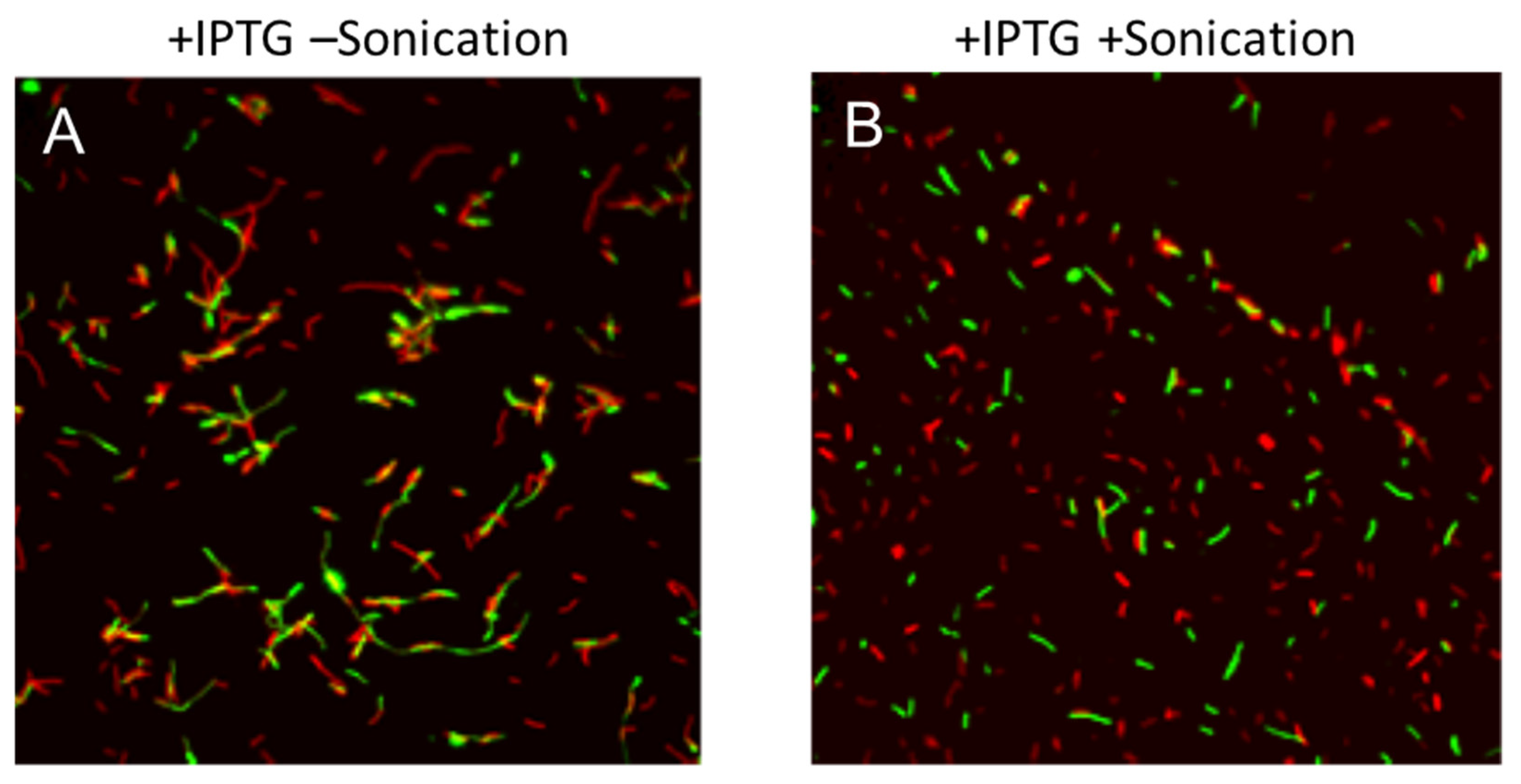
3.2. Microscopic Observation
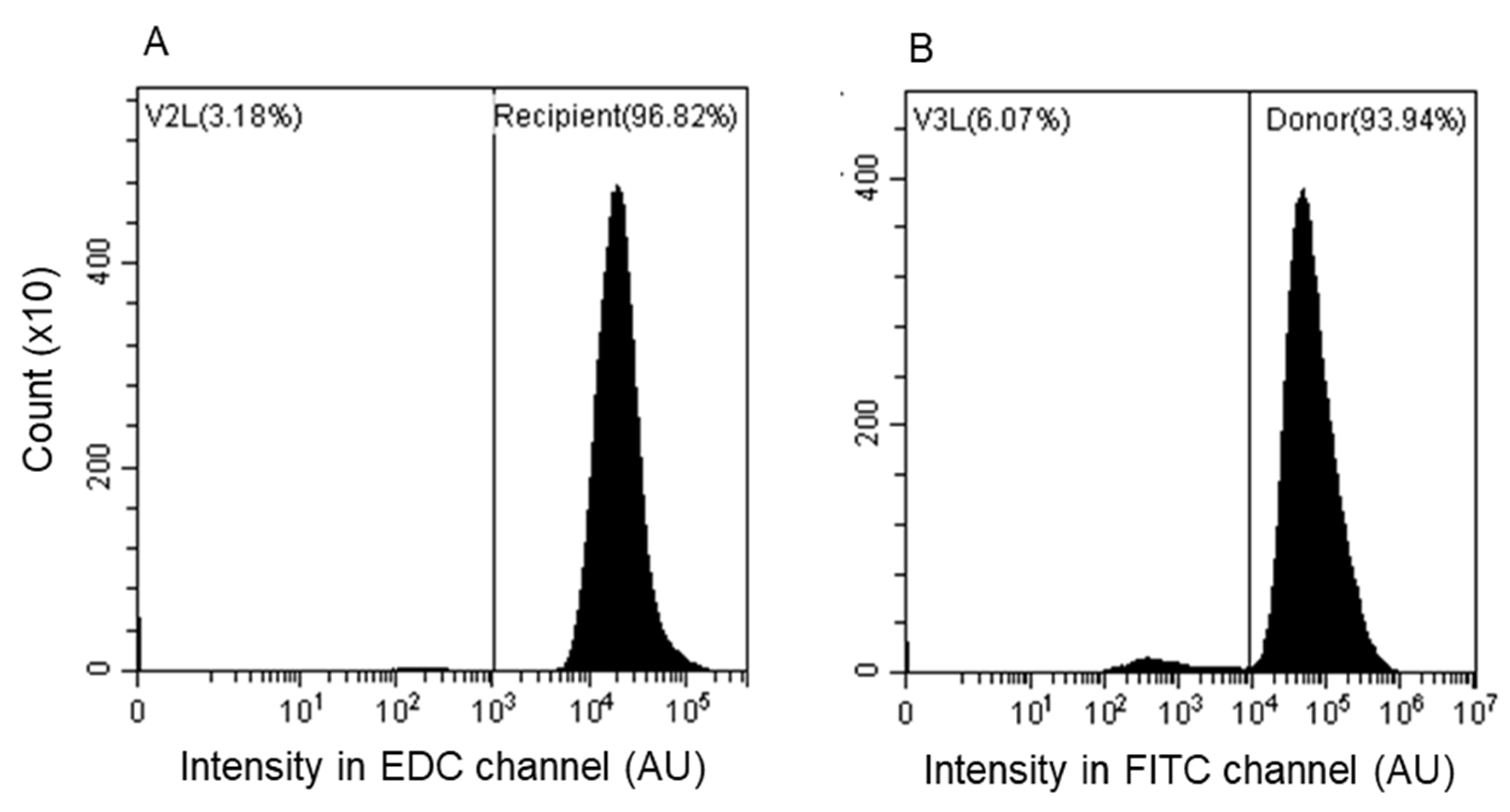
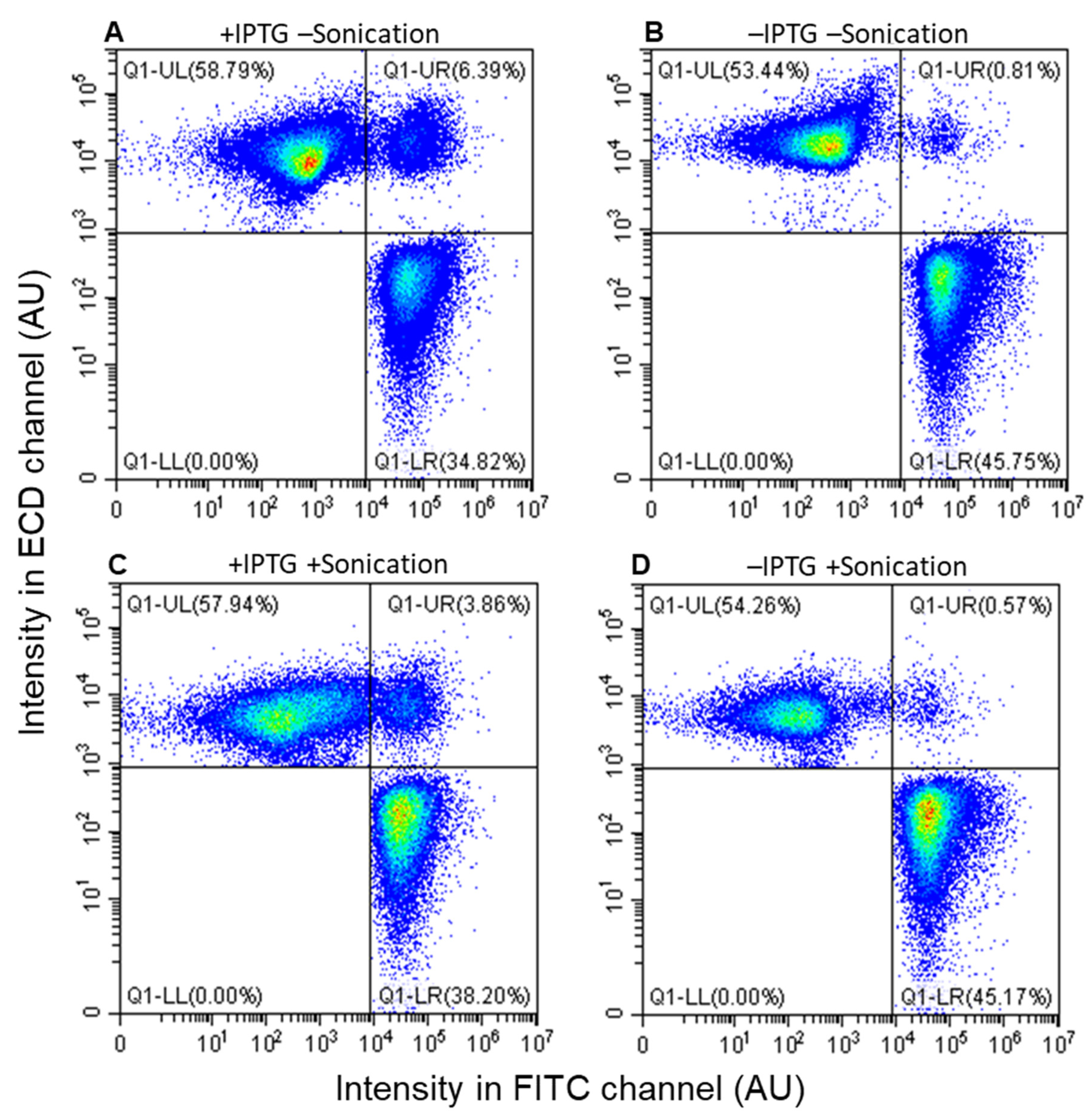
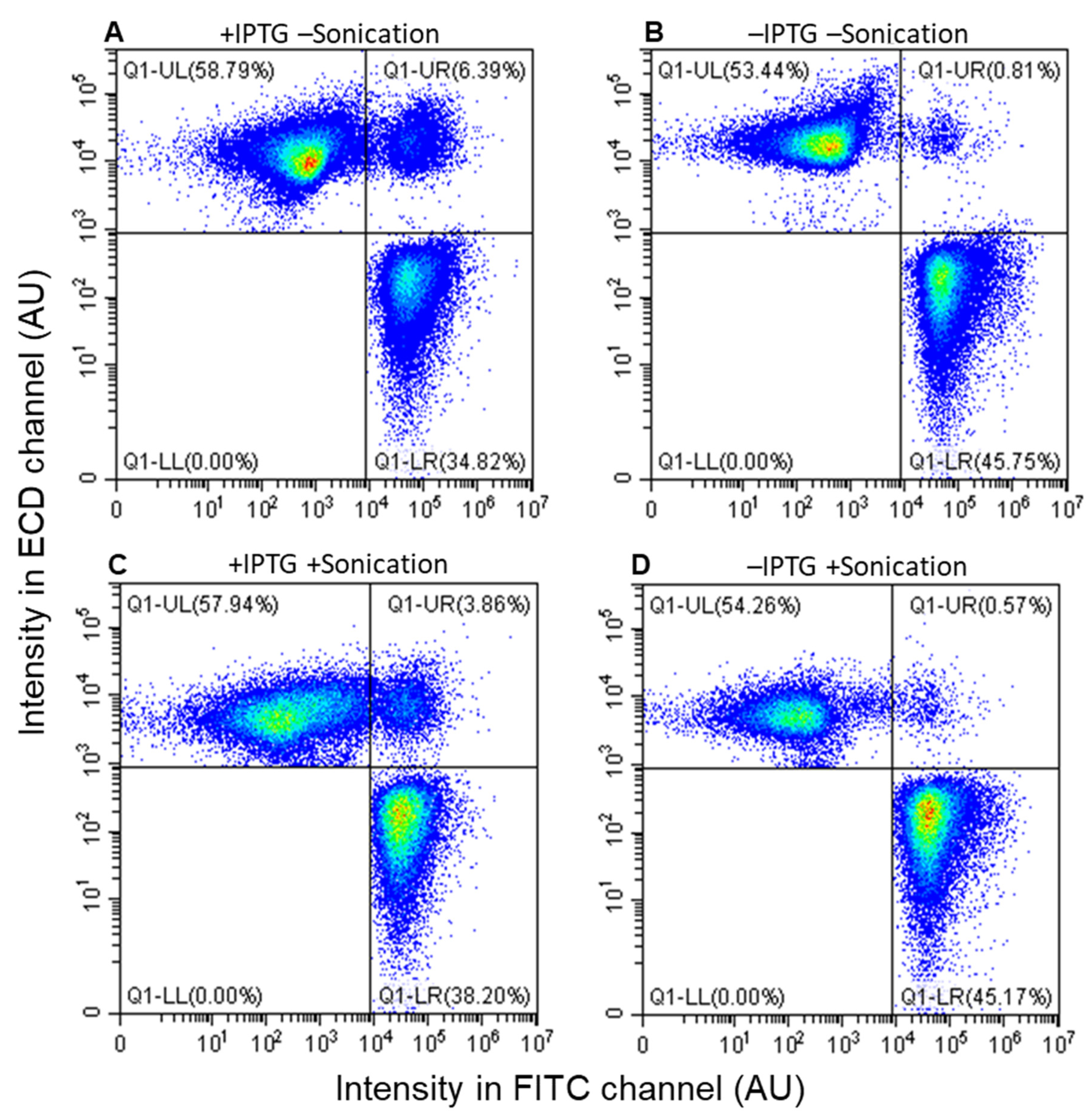
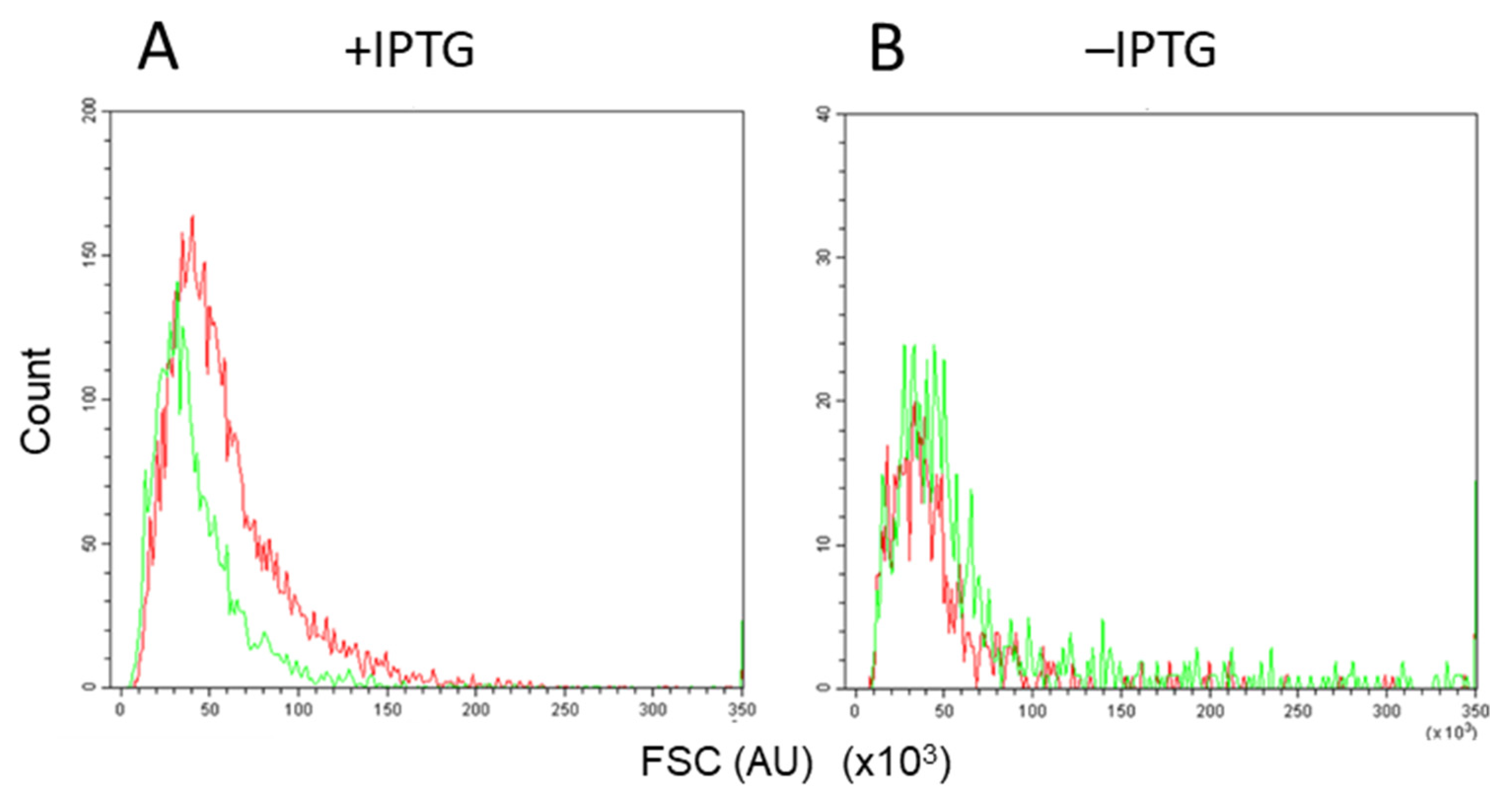
3.3. Assessment by Fluorescence-Activated Flow Cytometry
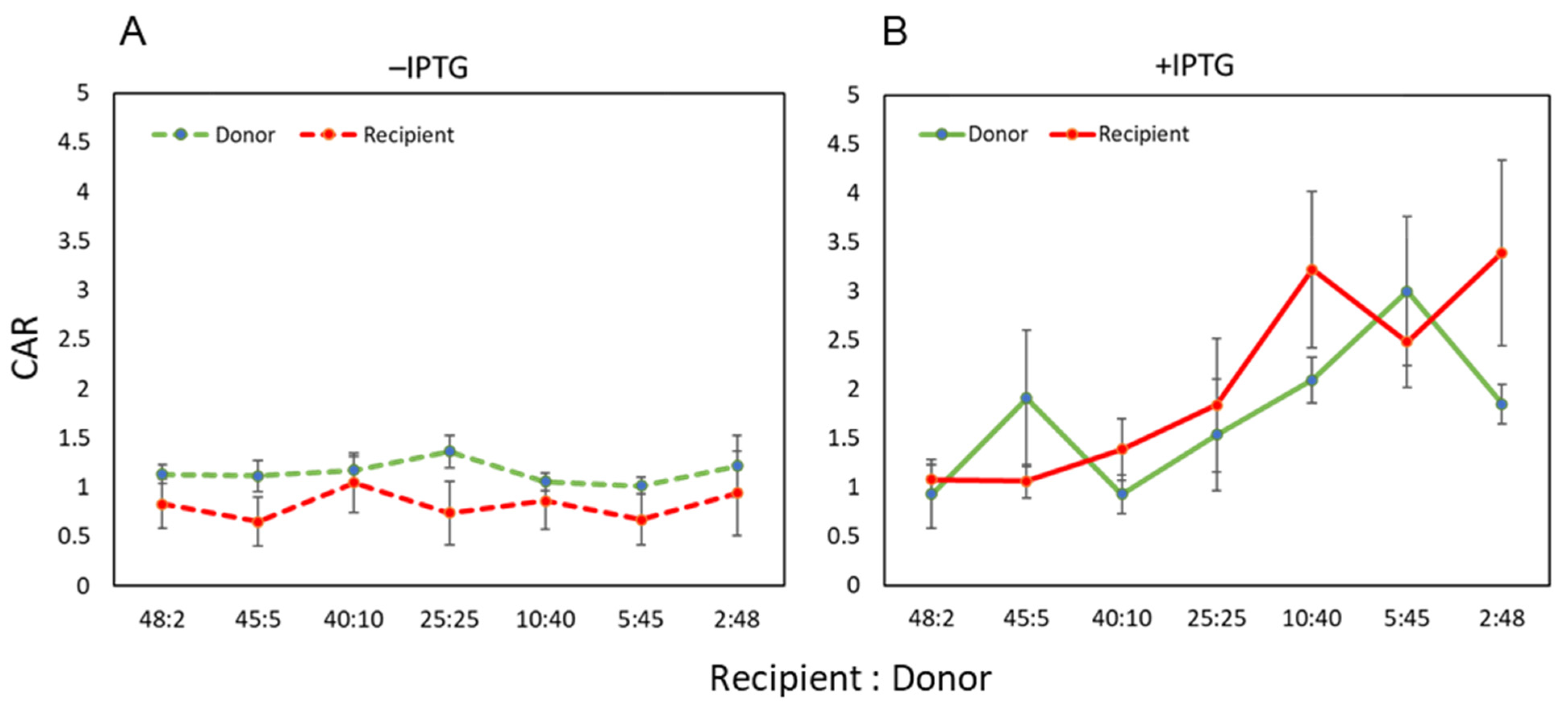
3.4. Accurate Assessment of Conjugation Efficiency
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Hu, B.; Khara, P.; Christie, P.J. Structural bases for F plasmid conjugation and F pilus biogenesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2019, 116, 14222–14227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquiod, S.; Brejnrod, A.; Morberg, S.M.; Abu Al-Soud, W.; Sørensen, S.J.; Riber, L. Deciphering conjugative plasmid permissiveness in wastewater microbiomes. Mol. Ecol. 2017, 26, 3556–3571. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Christie, P.J.; Dubnau, D. The ins and outs of DNA transfer in bacteria. Science 2005, 310, 1456–1460. [Google Scholar] [CrossRef] [Green Version]
- Arutyunov, D.; Frost, L.S. F conjugation: Back to the beginning. Plasmid 2013, 70, 18–32. [Google Scholar] [CrossRef]
- Cabezón, E.; Ripoll-Rozada, J.; Peña, A.; de la Cruz, F.; Arechaga, I. Towards an integrated model of bacterial conjugation. FEMS Microbiol. Rev. 2015, 39, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Goessweiner-Mohr, N.; Arends, K.; Keller, W.; Grohmann, E. Conjugation in Gram-Positive Bacteria. Microbiol. Spectr. 2014, 2, PLAS-0004-2013. [Google Scholar] [CrossRef] [Green Version]
- Koraimann, G. Spread and persistence of virulence and antibiotic resistance genes: A ride on the F plasmid conjugation module. EcoSal Plus 2018, 8, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.G.; Hu, B.; Christie, P.J. Biological and structural diversity of type IV secretion systems. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Waksman, G. From conjugation to T4S systems in Gram-negative bacteria: A mechanistic biology perspective. EMBO Rep. 2019, 20, e47012. [Google Scholar] [CrossRef] [PubMed]
- Christie, P.J.; Whitaker, N.; González-Rivera, C. Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 2014, 1843, 1578–1591. [Google Scholar] [CrossRef] [Green Version]
- Christie, P.J. The mosaic type IV secretion systems. EcoSal Plus 2016, 7, ESP-0020-2015. [Google Scholar] [CrossRef] [Green Version]
- Smillie, C.; Garcillán-Barcia, M.P.; Francia, M.V.; Rocha, E.P.; de la Cruz, F. Mobility of plasmids. Microbiol. Mol. Biol. Rev. 2010, 74, 434–452. [Google Scholar] [CrossRef] [Green Version]
- Baxter, J.C.; Funnell, B.E. Plasmid partition mechanisms. Microbiol. Spectr. 2014, 2, PLAS-0023-2014. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.C.; Hwang, L.C. Reconstitutions of plasmid partition systems and their mechanisms. Plasmid 2017, 91, 37–41. [Google Scholar] [CrossRef] [Green Version]
- Bouet, J.Y.; Funnell, B.E. Plasmid Localization and Partition in Enterobacteriaceae. EcoSal Plus 2019, 8, ESP-0003-2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonenshein, A.L.; Hoch, J.A.; Losick, R. Bacillus subtilis and Other Gram-Positive Bacteria: Biochemistry, Physiology, and Molecular Genetics; American Society for Microbiology Press: Washington, DC, USA, 1993. [Google Scholar] [CrossRef]
- Sonenshein, A.L.; Hoch, J.A.; Losick, R. Bacillus subtilis and Its Closest Relatives: From Genes to Cells; American Society for Microbiology Press: Washington, DC, USA, 2001. [Google Scholar] [CrossRef]
- Tanaka, T.; Koshikawa, T. Isolation and characterization of four types of plasmids from Bacillus subtilis (natto). J. Bacteriol. 1977, 131, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Koehler, T.M.; Thorne, C.B. Bacillus subtilis (natto) plasmid pLS20 mediates interspecies plasmid transfer. J. Bacteriol. 1987, 169, 5271–5278. [Google Scholar] [CrossRef] [Green Version]
- Meijer, W.J.; Wisman, G.B.; Terpstra, P.; Thorsted, P.B.; Thomas, C.M.; Holsappel, S.; Venema, G.; Bron, S. Rolling-circle plasmids from Bacillus subtilis: Complete nucleotide sequences and analyses of genes of pTA1015, pTA1040, pTA1050 and pTA1060, and comparisons with related plasmids from gram-positive bacteria. FEMS Microbiol. Rev. 1998, 21, 337–368. [Google Scholar] [CrossRef] [PubMed]
- Itaya, M.; Sakaya, N.; Matsunaga, S.; Fujita, K.; Kaneko, S. Conjugational transfer kinetics of pLS20 between Bacillus subtilis in liquid medium. Biosci. Biotechnol. Biochem. 2006, 70, 740–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.K.; Ramachandran, G.; Ramos-Ruiz, R.; Peiró-Pastor, R.; Abia, D.; Wu, L.J.; Meijer, W.J. Mobility of the native Bacillus subtilis conjugative plasmid pLS20 is regulated by intercellular signaling. PLoS Genet. 2013, 9, e1003892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Singh, P.K.; Luque-Ortega, J.R.; Yuste, L.; Alfonso, C.; Rojo, F.; Wu, L.J.; Meijer, W.J. A complex genetic switch involving overlapping divergent promoters and DNA looping regulates expression of conjugation genes of a gram-positive plasmid. PLoS Genet. 2014, 10, e1004733. [Google Scholar] [CrossRef] [Green Version]
- Meijer, W.J.J.; Boer, D.R.; Ares, S.; Alfonso, C.; Rojo, F.; Luque-Ortega, J.R.; Wu, L.J. Multiple layered control of the conjugation process of the Bacillus subtilis plasmid pLS20. Front. Mol. Biosci. 2021, 8, 648468. [Google Scholar] [CrossRef]
- Pinilla-Redondo, R.; Riber, L.; Sørensen, S.J. Fluorescence recovery allows the implementation of a fluorescence reporter gene platform applicable for the detection and quantification of horizontal gene transfer in anoxic environments. Appl. Environ. Microbiol. 2018, 84, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.R.; Hands, C.L.; Coello-Garcia, T.; Sani, B.S.; Ott, A.I.G.; Smith, S.J.; Davenport, R.J. A flow cytometry method for bacterial quantification and biomass estimates in activated sludge. J. Microbiol. Methods 2019, 160, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Klümper, U.; Riber, L.; Dechesne, A.; Sannazzarro, A.; Hansen, L.H.; Sørensen, S.J.; Smets, B.F. Broad host range plasmids can invade an unexpectedly diverse fraction of a soil bacterial community. ISME J. 2015, 9, 934–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinilla-Redondo, R.; Cyriaque, V.; Jacquiod, S.; Sørensen, S.J.; Riber, L. Monitoring plasmid-mediated horizontal gene transfer in microbiomes: Recent advances and future perspectives. Plasmid 2018, 99, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Conacher, C.G.; Naidoo-Blassoples, R.K.; Rossouw, D.; Bauer, F.F. Real-time monitoring of population dynamics and physical interactions in a synthetic yeast ecosystem by use of multicolour flow cytometry. Appl. Microbiol. Biotechnol. 2020, 104, 5547–5562. [Google Scholar] [CrossRef]
- Hellweger, F.L.; Clegg, R.J.; Clark, J.R.; Plugge, C.M.; Kreft, J.U. Advancing microbial sciences by individual-based modelling. Nat. Rev. Microbiol. 2016, 14, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Miyano, M.; Tanaka, K.; Ishikawa, S.; Takenaka, S.; Miguel-Arribas, A.; Meijer, W.J.J.; Yoshida, K. Rapid conjugative mobilization of a 100 kb segment of Bacillus subtilis chromosomal DNA is mediated by a helper plasmid with no ability for self-transfer. Microb. Cell Fact. 2018, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Miguel-Arribas, A.; Abia, D.; Singh, P.K.; Crespo, I.; Gago-Córdoba, C.; Hao, J.A.; Luque-Ortega, J.R.; Alfonso, C.; Wu, L.J.; et al. Discovery of a new family of relaxases in Firmicutes bacteria. PLoS Genet. 2017, 13, e1006586. [Google Scholar] [CrossRef]
- Guiziou, S.; Sauveplane, V.; Chang, H.J.; Clerté, C.; Declerck, N.; Jules, M.; Bonnet, J. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016, 44, 7495–7508. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, M.; Oshima, T.; Ogasawara, N. Involvement of the YneS/YgiH and PlsX proteins in phospholipid biosynthesis in both Bacillus subtilis and Escherichia coli. BMC Microbiol. 2007, 7, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, S.; Kawai, Y.; Hiramatsu, K.; Kuwano, M.; Ogasawara, N. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol. Microbiol. 2006, 60, 1364–1380. [Google Scholar] [CrossRef]
- Berka, R.M.; Hahn, J.; Albano, M.; Draskovic, I.; Persuh, M.; Cui, X.; Sloma, A.; Widner, W.; Dubnau, D. Microarray analysis of the Bacillus subtilis K-state: Genome-wide expression changes dependent on ComK. Mol. Microbiol. 2002, 43, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Pédelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a super folder green fluorescent protein. Nat. Biotechnol. 2005, 24, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Radeck, J.; Kraft, K.; Bartels, J.; Cikovic, T.; Durr, F.; Emenegger, J.; Kelterborn, S.; Sauer, C.; Fritz, G.; Gebhard, S.; et al. The Bacillus BioBrick Box: Generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J. Biol. Eng. 2013, 7, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Su, L.; Li, N.; Wu, J. Enhanced extracellular expression of Bacillus stearothermophilus α-amylase in Bacillus subtilis through signal peptide optimization, chaperone overexpression and α-amylase mutant selection. Microbial. Cell Fact. 2019, 18, 69. [Google Scholar] [CrossRef]
- Okumura, H.; Yoshimura, M.; Ueki, M.; Oshima, T.; Ogasawara, N.; Ishikawa, S. Regulation of chromosomal replication initiation by oriC-proximal DnaA-box clusters in Bacillus subtilis. Nucleic Acids Res. 2012, 40, 220–234. [Google Scholar] [CrossRef]
- Craig, L.; Forest, K.T.; Maier, B. Type IV pili: Dynamics, biophysics and functional consequences. Nat. Rev. Microbiol. 2019, 17, 429–440. [Google Scholar] [CrossRef]
- Gago-Córdoba, C.; Val-Calvo, J.; Abia, D.; Díaz-Talavera, A.; Miguel-Arribas, A.; Aguilar Suárez, R.; van Dijl, J.M.; Wu, L.J.; Meijer, W.J.J. A conserved class II type thioester domain-containing adhesin is required for efficient conjugation in Bacillus subtilis. mBio 2021, 12, e00104-21. [Google Scholar] [CrossRef]
- Ou, J.T. Role of surface exclusion genes in lethal zygosis in Escherichia coli K12 mating. Molec. Gen. Genet. 1980, 178, 573–581. [Google Scholar] [CrossRef]
- Kearns, D.B.; Losick, R. Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev. 2005, 19, 3083–3094. [Google Scholar] [CrossRef] [Green Version]
- Lardon, L.A.; Merkey, B.V.; Martins, S.; Dötsch, A.; Picioreanu, C.; Kreft, J.U.; Smets, B.F. iDynoMiCS: Next-generation individual-based modelling of biofilms. Environ. Microbiol. 2011, 13, 2416–2434. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Description | Source or Reference |
|---|---|---|
| E. coli | ||
| C600 | Plasmid cloning host | Laboratory stock |
| B. subtilis | ||
| 168 | trpC2 | Laboratory stock |
| YNB102 | trpC2, amyE::(Pspank-rappLS20 spc), pLS20catΔoriT | [31] |
| KV7 | trpC2, ΔcomK::tet | This study |
| KV9 | trpC2, amyE::(Pspank-rappLS20 spc), pGR16B, pLS20catΔoriT | This study |
| KV10 | trpC2, ΔcomK::tet, aprE::(PrpsO-mKATE2 kan) | This study |
| KV11 | trpC2, amyE::(Pspank-rappLS20 spec), pGR16B_sfGFP, pLS20catΔoriT | This study |
| Plasmid | ||
| pLS20catΔoriT | pLS20cat lacking the oriT region | [31] |
| pGR16B | erm, amp | [32] |
| pGR16B_sfGFP | pGR16B carrying sfGFP | This study |
| pDG-SG51 | mKATE2 | [33] |
| pANPCK | kan | [34] |
| pOGW | tet | [35] |
| Primer | Sequence (5′-3′) |
|---|---|
| aprE_D_F | cggaaggatactacatcctggttaatcaacgtacaagcagctgcac |
| aprE_D_R | ggccgagcagtattcgaatgtcaag |
| aprEU_F3_nested | caccgagctcatagcttgtcgcgatcacctcatcc |
| aprE_D_R2_nested | tgctttcgctgattacaacattggtgacgctgcct |
| aprE_U_F | ccggtacttgccaccacatcataac |
| aprE_U_PrpsO_R | aatttgcgtgcgttgcaagttatttccgcactctcgctatttccgtagagactcg |
| comK_up_F | tgaaggattggcttattcgctctgc |
| comK_up_R | cagtatttcatcacttatacaacaactaataatctatcatctgtttttg |
| comK_Down_F | gcctggcagttccctactctcgcatgcgtgagctcggggaacggtattag |
| comK_Down_R | atcgaagatctgcctactgaacaaatc |
| comK_nested_F | gcttgagcgctgcatattctttagagagcg |
| comK_nested_R | gttgtaaaagcggcgcttccgtatttgccg |
| Kan_F | ggatagactccaccagaagagccgcaagcttacgataaacccagc |
| Kan_terminator_R | ccaggatgtagtatccttccgaaaaaatcccgccgctggcgggattttaactaggtactaaaacaattcatcc |
| mKate2_F | gggctaaatatgatttggaggtgaaacaggatgtcagaactaatcaaagagaatatg |
| mKate2_Kan_R | gctcttctggtggagtctatcctataaacgcagaaaggcccacccgaag |
| tetR_F | ttgttgtataagtgatgaaatactg |
| tetR_R | atgcgagagtagggaactgccaggc |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, K.; Verrone, V.; Amatsu, R.; Fukui, K.; Meijer, W.J.J.; Ishikawa, S.; Wipat, A.; Yoshida, K.-i. Assessment of Bacillus subtilis Plasmid pLS20 Conjugation in the Absence of Quorum Sensing Repression. Microorganisms 2021, 9, 1931. https://doi.org/10.3390/microorganisms9091931
Mori K, Verrone V, Amatsu R, Fukui K, Meijer WJJ, Ishikawa S, Wipat A, Yoshida K-i. Assessment of Bacillus subtilis Plasmid pLS20 Conjugation in the Absence of Quorum Sensing Repression. Microorganisms. 2021; 9(9):1931. https://doi.org/10.3390/microorganisms9091931
Chicago/Turabian StyleMori, Kotaro, Valeria Verrone, Ryotaro Amatsu, Kaho Fukui, Wilfried J. J. Meijer, Shu Ishikawa, Anil Wipat, and Ken-ichi Yoshida. 2021. "Assessment of Bacillus subtilis Plasmid pLS20 Conjugation in the Absence of Quorum Sensing Repression" Microorganisms 9, no. 9: 1931. https://doi.org/10.3390/microorganisms9091931
APA StyleMori, K., Verrone, V., Amatsu, R., Fukui, K., Meijer, W. J. J., Ishikawa, S., Wipat, A., & Yoshida, K.-i. (2021). Assessment of Bacillus subtilis Plasmid pLS20 Conjugation in the Absence of Quorum Sensing Repression. Microorganisms, 9(9), 1931. https://doi.org/10.3390/microorganisms9091931