
Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
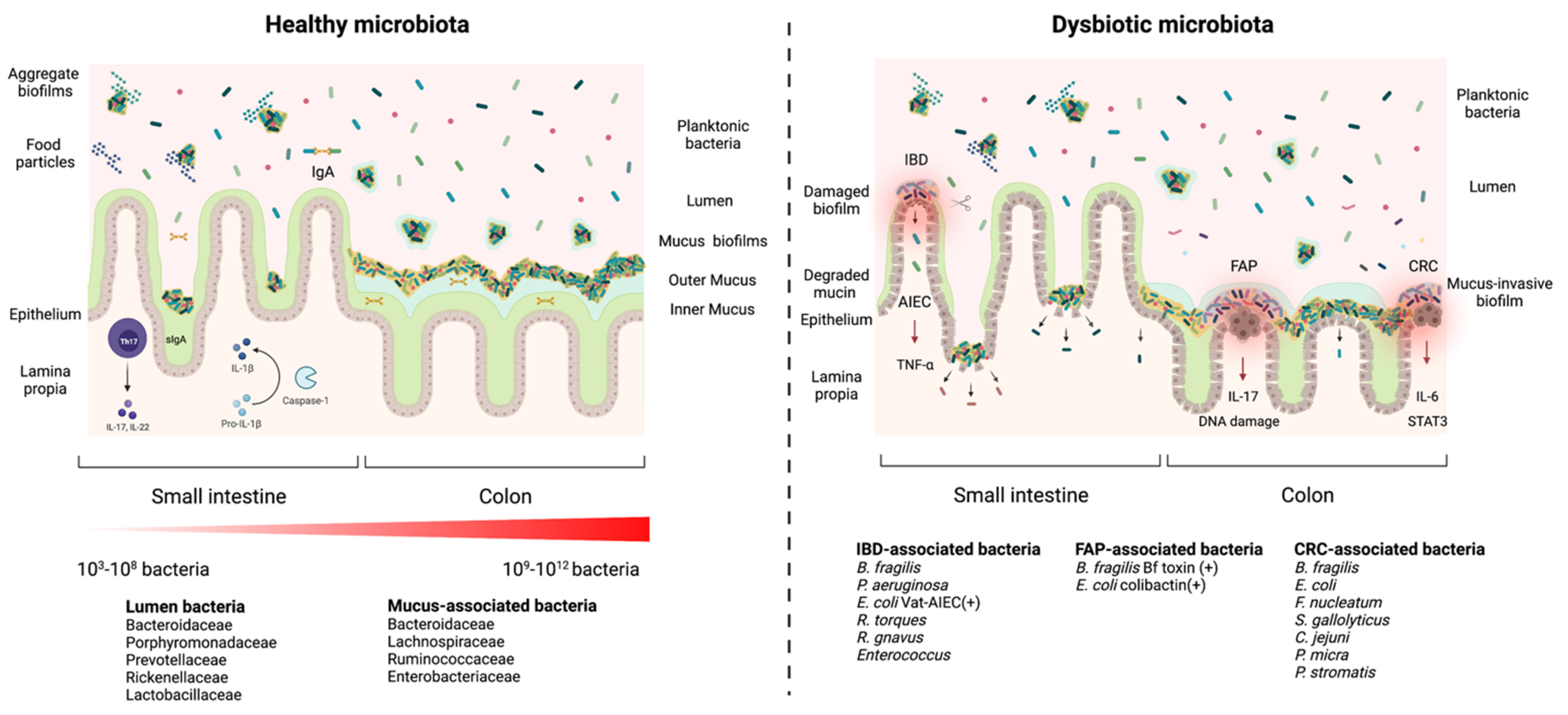
2. Biofilm Formation in the Gastrointestinal Tract: The Blurry Line between Health and Disease
3. Gut Biofilm Communities: Location, Organization, and Composition
4. Health and Disease: Non-Invasive versus Invasive Gut Microbial Biofilms
5. Diversity of Interactions and Phenotypes in the Gut Biofilm Communities
5.1. Competitive and Cooperative Behaviors in a Biofilm
5.2. Phenotypic and Metabolic Heterogeneity Inside a Biofilm
6. Diverse Gut and Microbiota-Derived Signals Induce Biofilm Formation in Commensal Bacteria and Enteropathogens
6.1. Host-Derived Factors and Biofilm Formation
6.2. Antibiotics Affecting Biofilm Formation
6.3. Microbiota Metabolites and Biofilm Formation
6.4. Bacterial and Phages Interactions Affect Biofilm Formation
7. The Case for C. difficile
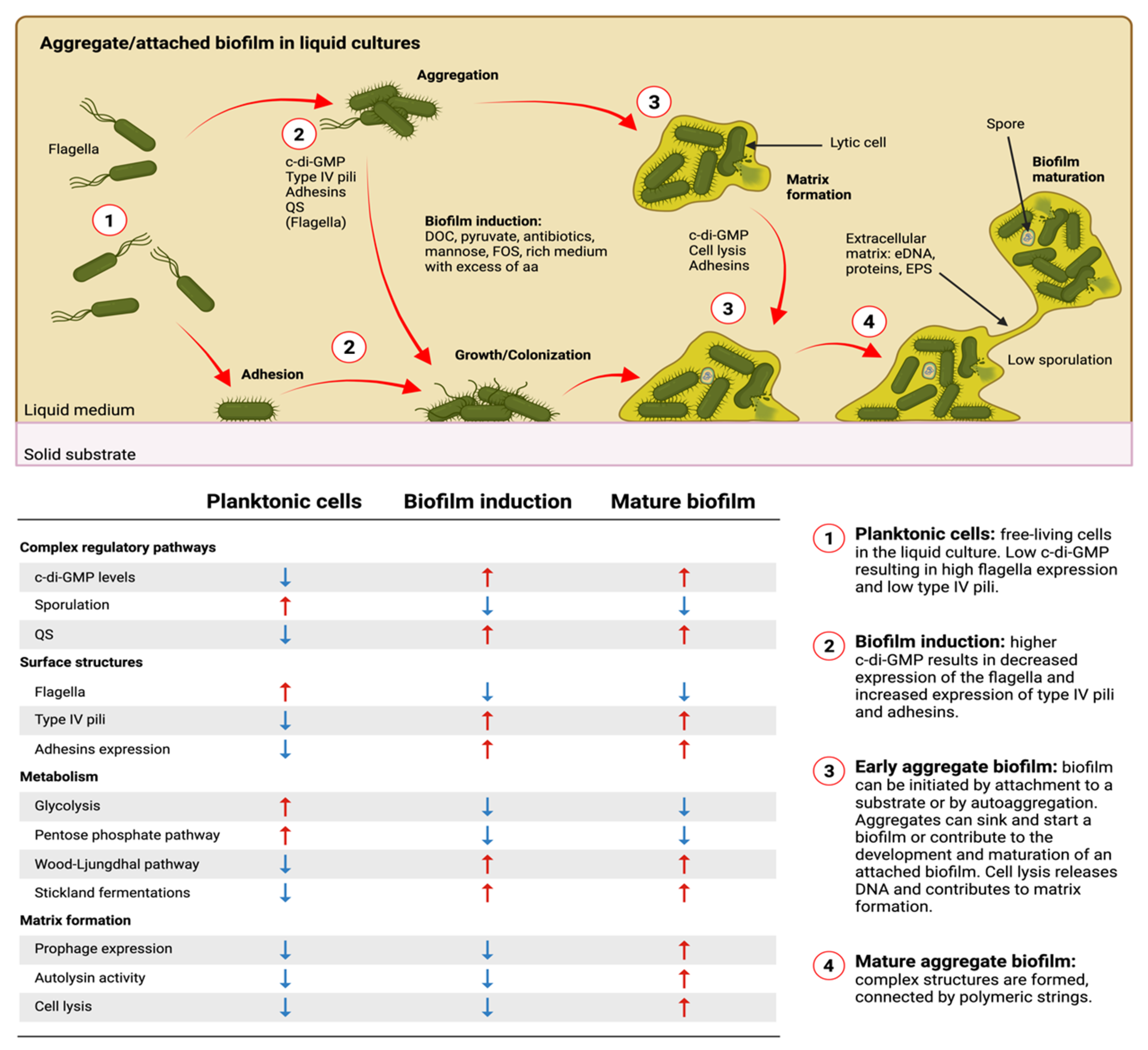
8. Biofilm Formation in C. difficile
8.1. In Vitro Models, Quantification, and Visualization
8.2. In Vivo Models and Clinical Data
8.3. Composition of the In Vitro Biofilm Matrix
8.4. How Is eDNA Released into the Biofilm Matrix?
8.5. Surface Structures and Their Importance in Biofilm Formation
9. Regulation of Biofilm Formation
9.1. Are SinR and SinR’ Involved?
9.2. Is Quorum Sensing Important for Biofilm Formation?
9.3. The Important Role of c-di-GMP in C. difficile Biofilm Formation
9.4. Post-Transcriptional Regulation and Phenotypic Heterogeneity in Biofilms
10. What Induces Biofilm Formation?
10.1. Induction of Biofilm Formation by Antibiotics
10.2. Induction of Biofilm Formation by DOC
11. The Importance of Metabolism in Biofilm Formation
12. Role of the Microbiota-C. difficile Interactions in Biofilm Formation
13. Gut Biofilm: A Shelter against Stresses for C. difficile
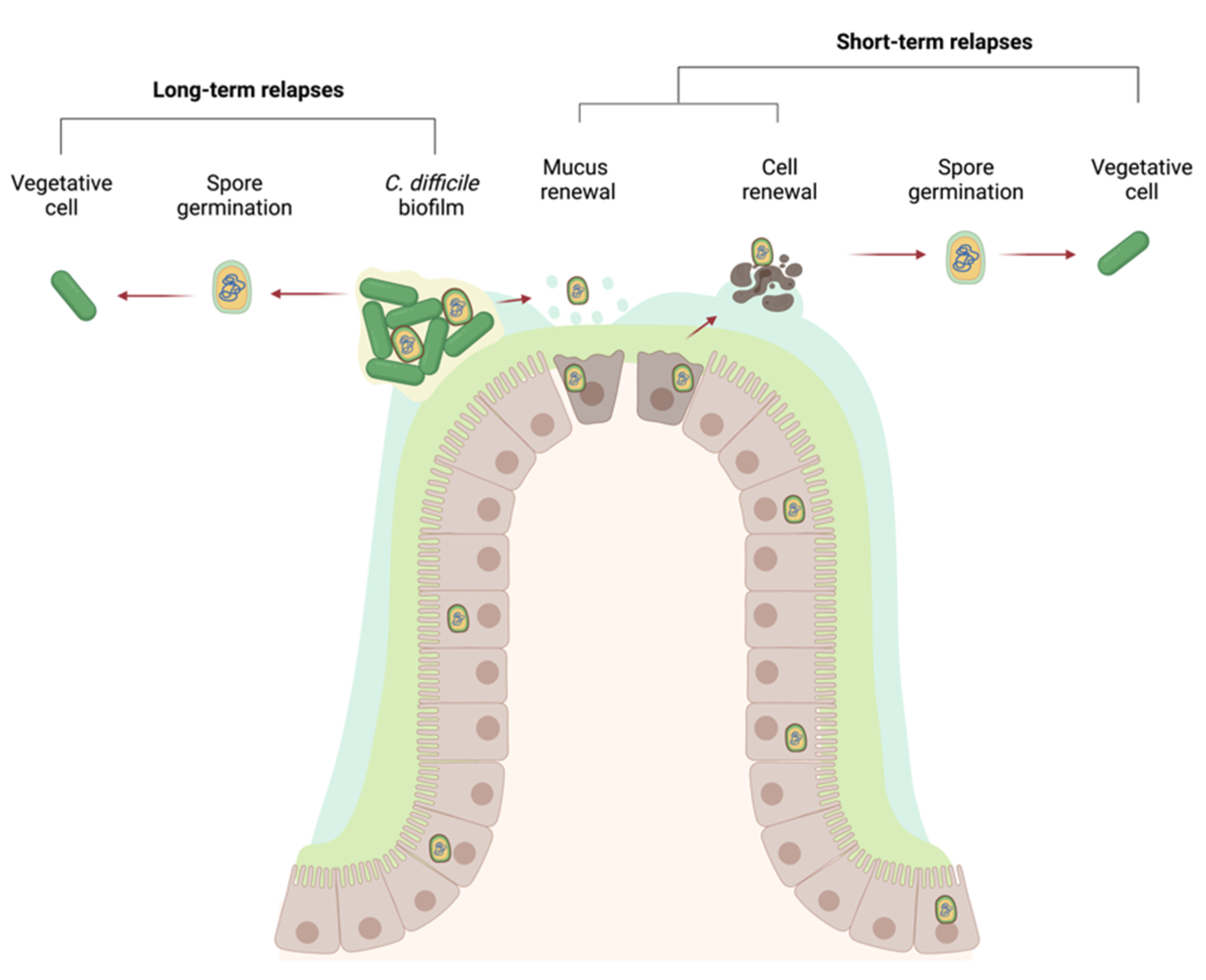
14. Persistence in the Gut: Spores, Biofilms, or Both?
15. Refining the Infectious Cycle of C. difficile: Metabolic Landscape as a Determinant of Biofilm Formation, Pathogenesis or Sporulation
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eckburg, P.B. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How Host-Microbial Interactions Shape the Nutrient Environment of the Mammalian Intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef]
- Hooper, L.V.; Wong, M.H.; Thelin, A.; Hansson, L.; Falk, P.G.; Gordon, J.I. Molecular Analysis of Commensal Host-Microbial Relationships in the Intestine. Science 2001, 291, 881–884. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, Stability and Resilience of the Human Gut Microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef]
- Motta, J.-P.; Wallace, J.L.; Buret, A.G.; Deraison, C.; Vergnolle, N. Gastrointestinal Biofilms in Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 314–334. [Google Scholar] [CrossRef]
- Stoodley, P.; Sauer, K.; Davies, D.G.; Costerton, J.W. Biofilms as Complex Differentiated Communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef]
- Guzmán-Soto, I.; McTiernan, C.; Gonzalez-Gomez, M.; Ross, A.; Gupta, K.; Suuronen, E.J.; Mah, T.-F.; Griffith, M.; Alarcon, E.I. Mimicking Biofilm Formation and Development: Recent Progress in in vitro and in Vivo Biofilm Models. Iscience 2021, 24, 102443. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial Biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The Biofilm Matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Bollinger, R.R.; Barbas, A.S.; Bush, E.L.; Lin, S.S.; Parker, W. Biofilms in the Normal Human Large Bowel: Fact Rather than Fiction. Gut 2007, 56, 1481–1482. [Google Scholar]
- Macfarlane, S.; Dillon, J.F. Microbial Biofilms in the Human Gastrointestinal Tract. J. Appl. Microbiol. 2007, 102, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Sonnenburg, J.L.; Angenent, L.T.; Gordon, J.I. Getting a Grip on Things: How Do Communities of Bacterial Symbionts Become Established in Our Intestine? Nat. Immunol. 2004, 5, 569–573. [Google Scholar] [CrossRef]
- Tropini, C.; Earle, K.A.; Huang, K.C.; Sonnenburg, J.L. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe 2017, 21, 433–442. [Google Scholar] [CrossRef] [PubMed]
- von Rosenvinge, E.C.; O’May, G.A.; Macfarlane, S.; Macfarlane, G.T.; Shirtliff, M.E. Microbial Biofilms and Gastrointestinal Diseases. Pathog. Dis. 2013, 67, 25–38. [Google Scholar] [CrossRef]
- Macfarlane, S.; Furrie, E.; Macfarlane, G.T.; Dillon, J.F. Microbial Colonization of the Upper Gastrointestinal Tract in Patients with Barrett’s Esophagus. Clin. Infect. Dis. 2007, 45, 29–38. [Google Scholar] [CrossRef]
- Macfarlane, S.; Furrie, E.; Cummings, J.H.; Macfarlane, G.T. Chemotaxonomic Analysis of Bacterial Populations Colonizing the Rectal Mucosa in Patients with Ulcerative Colitis. Clin. Infect. Dis. 2004, 38, 1690–1699. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Herber, A. Mucosal Flora in Crohn’s Disease and Ulcerative Colitis—An Overview. J. Physiol. Pharmacol. 2009, 60 (Suppl. S6), 61–71. [Google Scholar]
- Coticchia, J.M.; Sugawa, C.; Tran, V.R.; Gurrola, J.; Kowalski, E.; Carron, M.A. Presence and Density of Helicobacter pylori Biofilms in Human Gastric Mucosa in Patients with Peptic Ulcer Disease. J. Gastrointest. Surg. 2006, 10, 883–889. [Google Scholar] [CrossRef]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota Organization Is a Distinct Feature of Proximal Colorectal Cancers. Proc. Natl. Acad Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef]
- Li, S.; Konstantinov, S.R.; Smits, R.; Peppelenbosch, M.P. Bacterial Biofilms in Colorectal Cancer Initiation and Progression. Trends Mol. Med. 2017, 23, 18–30. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef]
- Yang, J.; Yang, Y.; Ishii, M.; Nagata, M.; Aw, W.; Obana, N.; Tomita, M.; Nomura, N.; Fukuda, S. Does the Gut Microbiota Modulate Host Physiology through Polymicrobial Biofilms? Microb. Environ. 2020, 35, ME20037. [Google Scholar] [CrossRef]
- Wang, Y.; Antonopoulos, D.A.; Zhu, X.; Harrell, L.; Hanan, I.; Alverdy, J.C.; Meyer, F.; Musch, M.W.; Young, V.B.; Chang, E.B. Laser Capture Microdissection and Metagenomic Analysis of Intact Mucosa-Associated Microbial Communities of Human Colon. Appl. Microbiol. Biotechnol. 2010, 88, 1333–1342. [Google Scholar] [CrossRef][Green Version]
- Hooper, L.V.; Gordon, J.I. Commensal Host-Bacterial Relationships in the Gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- de Vos, W.M. Microbial Biofilms and the Human Intestinal Microbiome. NPJ Biofilms Microbiomes 2015, 1, 15005. [Google Scholar] [CrossRef]
- Engevik, M.A.; Danhof, H.A.; Auchtung, J.; Endres, B.T.; Ruan, W.; Bassères, E.; Engevik, A.C.; Wu, Q.; Nicholson, M.; Luna, R.A.; et al. Fusobacterium nucleatum Adheres to Clostridioides difficile via the RadD Adhesin to Enhance Biofilm Formation in Intestinal Mucus. Gastroenterology 2021, 160, 1301–1314.e8. [Google Scholar] [CrossRef] [PubMed]
- Lebeer, S.; Verhoeven, T.L.A.; Claes, I.J.J.; De Hertogh, G.; Vermeire, S.; Buyse, J.; Van Immerseel, F.; Vanderleyden, J.; De Keersmaecker, S.C.J. FISH Analysis of Lactobacillus Biofilms in the Gastrointestinal Tract of Different Hosts. Lett Appl. Microbiol. 2011, 52, 220–226. [Google Scholar] [CrossRef]
- Nava, G.M.; Friedrichsen, H.J.; Stappenbeck, T.S. Spatial Organization of Intestinal Microbiota in the Mouse Ascending Colon. ISME J. 2011, 5, 627–638. [Google Scholar] [CrossRef]
- Palestrant, D.; Holzknecht, Z.E.; Collins, B.H.; Parker, W.; Miller, S.E.; Bollinger, R.R. Microbial Biofilms in the Gut: Visualization by Electron Microscopy and by Acridine Orange Staining. Ultrastruct. Pathol. 2004, 28, 23–27. [Google Scholar] [CrossRef]
- Smith, H.F.; Fisher, R.E.; Everett, M.L.; Thomas, A.D.; Randal Bollinger, R.; Parker, W. Comparative Anatomy and Phylogenetic Distribution of the Mammalian Cecal Appendix. J. Evol. Biol. 2009, 22, 1984–1999. [Google Scholar] [CrossRef]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Oh, K.; Ren, B.; Tickle, T.L.; Franzosa, E.A.; Wachtman, L.M.; Miller, A.D.; Westmoreland, S.V.; Mansfield, K.G.; Vallender, E.J.; et al. Biogeography of the Intestinal Mucosal and Lumenal Microbiome in the Rhesus Macaque. Cell Host Microbe 2015, 17, 385–391. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; von Wright, A.; Vilpponen-Salmela, T.; Ben-Amor, K.; Akkermans, A.D.L.; de Vos, W.M. Mucosa-Associated Bacteria in the Human Gastrointestinal Tract Are Uniformly Distributed along the Colon and Differ from the Community Recovered from Feces. Appl. Environ. Microbiol. 2002, 68, 3401–3407. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Larsson, J.M.H.; Hansson, G.C. The Two Mucus Layers of Colon Are Organized by the MUC2 Mucin, Whereas the Outer Layer Is a Legislator of Host-Microbial Interactions. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4659–4665. [Google Scholar] [CrossRef]
- Gustafsson, J.K.; Ermund, A.; Johansson, M.E.V.; Schütte, A.; Hansson, G.C.; Sjövall, H. An Ex Vivo Method for Studying Mucus Formation, Properties, and Thickness in Human Colonic Biopsies and Mouse Small and Large Intestinal Explants. Am. J. Physiol.-Gastrointest. Liver Physiol. 2012, 302, G430–G438. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The Inner of the Two Muc2 Mucin-Dependent Mucus Layers in Colon Is Devoid of Bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef]
- Tailford, L.E.; Crost, E.H.; Kavanaugh, D.; Juge, N. Mucin Glycan Foraging in the Human Gut Microbiome. Front. Genet. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Douillard, F.P.; Ribbera, A.; Kant, R.; Pietilä, T.E.; Järvinen, H.M.; Messing, M.; Randazzo, C.L.; Paulin, L.; Laine, P.; Ritari, J.; et al. Comparative Genomic and Functional Analysis of 100 Lactobacillus rhamnosus Strains and Their Comparison with Strain GG. PLoS Genet. 2013, 9, e1003683. [Google Scholar] [CrossRef]
- Macfarlane, S.; McBain, A.J.; Macfarlane, G.T. Consequences of Biofilm and Sessile Growth in the Large Intestine. Adv. Dent. Res. 1997, 11, 59–68. [Google Scholar] [CrossRef]
- Bergstrom, K.; Shan, X.; Casero, D.; Batushansky, A.; Lagishetty, V.; Jacobs, J.P.; Hoover, C.; Kondo, Y.; Shao, B.; Gao, L.; et al. Proximal Colon–Derived O-Glycosylated Mucus Encapsulates and Modulates the Microbiota. Science 2020, 370, 467–472. [Google Scholar] [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of Pathogens and Pathobionts by the Gut Microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria Can Protect from Enteropathogenic Infection through Production of Acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Hammami, R.; Fernandez, B.; Lacroix, C.; Fliss, I. Anti-Infective Properties of Bacteriocins: An Update. Cell. Mol. Life Sci. 2013, 70, 2947–2967. [Google Scholar] [CrossRef]
- McDonald, J.A.K.; Mullish, B.H.; Pechlivanis, A.; Liu, Z.; Brignardello, J.; Kao, D.; Holmes, E.; Li, J.V.; Clarke, T.B.; Thursz, M.R.; et al. Inhibiting Growth of Clostridioides difficile by Restoring Valerate, Produced by the Intestinal Microbiota. Gastroenterology 2018, 155, 1495–1507.e15. [Google Scholar] [CrossRef]
- Macpherson, A.J.; Geuking, M.B.; McCoy, K.D. Homeland Security: IgA Immunity at the Frontiers of the Body. Trends Immunol. 2012, 33, 160–167. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The Antibacterial Lectin RegIIIgamma Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth Cells Directly Sense Gut Commensals and Maintain Homeostasis at the Intestinal Host-Microbial Interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef]
- Wrzosek, L.; Miquel, S.; Noordine, M.-L.; Bouet, S.; Joncquel Chevalier-Curt, M.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii Influence the Production of Mucus Glycans and the Development of Goblet Cells in the Colonic Epithelium of a Gnotobiotic Model Rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Kamada, N.; Nakamura, Y.; Burberry, A.; Kuffa, P.; Suzuki, S.; Shaw, M.H.; Kim, Y.-G.; Núñez, G. NLRC4-Driven Production of IL-1β Discriminates between Pathogenic and Commensal Bacteria and Promotes Host Intestinal Defense. Nat. Immunol. 2012, 13, 449–456. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Desai, M.S.; Seekatz, A.M.; Koropatkin, N.M.; Kamada, N.; Hickey, C.A.; Wolter, M.; Pudlo, N.A.; Kitamoto, S.; Terrapon, N.; Muller, A.; et al. A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility. Cell 2016, 167, 1339–1353.e21. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.W.; Keeney, K.M.; Crepin, V.F.; Rathinam, V.A.K.; Fitzgerald, K.A.; Finlay, B.B.; Frankel, G. Citrobacter rodentium: Infection, Inflammation and the Microbiota. Nat. Rev. Microbiol. 2014, 12, 612–623. [Google Scholar] [CrossRef]
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter pylori Activates and Expands Lgr5(+) Stem Cells Through Direct Colonization of the Gastric Glands. Gastroenterology 2015, 148, 1392–1404.e21. [Google Scholar] [CrossRef]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with Familial Adenomatous polyposis Harbor Colonic Biofilms Containing Tumorigenic Bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef]
- Chung, L.; Thiele Orberg, E.; Geis, A.L.; Chan, J.L.; Fu, K.; DeStefano Shields, C.E.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-Carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 203–214.e5. [Google Scholar] [CrossRef]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Herold, J.L.; Schady, D.; Davis, J.; Kopetz, S.; Martinez-Moczygemba, M.; Murray, B.E.; Han, F.; Li, Y.; Callaway, E.; et al. Streptococcus gallolyticus Subsp. Gallolyticus Promotes Colorectal Tumor Development. PLoS Pathog. 2017, 13, e1006440. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via Its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-Resolution Bacterial 16S RRNA Gene Profile Meta-Analysis and Biofilm Status Reveal Common Colorectal Cancer Consortia. NPJ Biofilms Microbiomes 2017, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-Occurrence of Anaerobic Bacteria in Colorectal Carcinomas. Microbiome 2013, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial Organization and Composition of the Mucosal Flora in Patients with Inflammatory Bowel Disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Molin, G.; Ahrné, S.; Adawi, D.; Jeppsson, B. High Proportions of Proinflammatory Bacteria on the Colonic Mucosa in a Young Patient with Ulcerative Colitis as Revealed by Cloning and Sequencing of 16S RRNA Genes. Dig. Dis. Sci. 2007, 52, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Rolhion, N.; Darfeuille-Michaud, A. Adherent-Invasive Escherichia coli in Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2007, 13, 1277–1283. [Google Scholar] [CrossRef]
- Gibold, L.; Garenaux, E.; Dalmasso, G.; Gallucci, C.; Cia, D.; Mottet-Auselo, B.; Faïs, T.; Darfeuille-Michaud, A.; Nguyen, H.T.T.; Barnich, N.; et al. The Vat-AIEC Protease Promotes Crossing of the Intestinal Mucus Layer by Crohn’s Disease-Associated Escherichia coli. Cell Microbiol. 2016, 18, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic Bacteria with Increased Prevalence in IBD Mucosa Augment in vitro Utilization of Mucin by Other Bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef]
- Golińska, E.; Tomusiak, A.; Gosiewski, T.; Więcek, G.; Machul, A.; Mikołajczyk, D.; Bulanda, M.; Heczko, P.B.; Strus, M. Virulence Factors of Enterococcus Strains Isolated from Patients with Inflammatory Bowel Disease. World J. Gastroenterol. 2013, 19, 3562–3572. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Rakoff-Nahoum, S. Understanding Competition and Cooperation within the Mammalian Gut Microbiome. Curr. Biol. 2019, 29, R538–R544. [Google Scholar] [CrossRef]
- Mitri, S.; Richard Foster, K. The Genotypic View of Social Interactions in Microbial Communities. Annu. Rev. Genet. 2013, 47, 247–273. [Google Scholar] [CrossRef]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica Serovar Typhimurium Exploits Inflammation to Compete with the Intestinal Microbiota. PLoS Biol. 2007, 5, e244. [Google Scholar] [CrossRef]
- Roelofs, K.G.; Coyne, M.J.; Gentyala, R.R.; Chatzidaki-Livanis, M.; Comstock, L.E. Bacteroidales Secreted Antimicrobial Proteins Target Surface Molecules Necessary for Gut Colonization and Mediate Competition In Vivo. mBio 2016, 7, e01055-16. [Google Scholar] [CrossRef]
- Aoki, S.K.; Pamma, R.; Hernday, A.D.; Bickham, J.E.; Braaten, B.A.; Low, D.A. Contact-Dependent Inhibition of Growth in Escherichia coli. Science 2005, 309, 1245–1248. [Google Scholar] [CrossRef]
- Sana, T.G.; Flaugnatti, N.; Lugo, K.A.; Lam, L.H.; Jacobson, A.; Baylot, V.; Durand, E.; Journet, L.; Cascales, E.; Monack, D.M. Salmonella typhimurium Utilizes a T6SS-Mediated Antibacterial Weapon to Establish in the Host Gut. Proc. Natl. Acad. Sci. USA 2016, 113, E5044–E5051. [Google Scholar] [CrossRef] [PubMed]
- Whitney, J.C.; Peterson, S.B.; Kim, J.; Pazos, M.; Verster, A.J.; Radey, M.C.; Kulasekara, H.D.; Ching, M.Q.; Bullen, N.P.; Bryant, D.; et al. A Broadly Distributed Toxin Family Mediates Contact-Dependent Antagonism between Gram-Positive Bacteria. Elife 2017, 6, e26938. [Google Scholar] [CrossRef] [PubMed]
- Nadell, C.D.; Drescher, K.; Foster, K.R. Spatial Structure, Cooperation and Competition in Biofilms. Nat. Rev. Microbiol. 2016, 14, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.; McNally, L.; Darch, S.E.; Brown, S.P.; Whiteley, M. The Biogeography of Polymicrobial Infection. Nat. Rev. Microbiol. 2016, 14, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Lion, S.; van Baalen, M. Self-Structuring in Spatial Evolutionary Ecology. Ecol. Lett. 2008, 11, 277–295. [Google Scholar] [CrossRef]
- Foster, K.R.; Bell, T. Competition, Not Cooperation, Dominates Interactions among Culturable Microbial Species. Curr. Biol. 2012, 22, 1845–1850. [Google Scholar] [CrossRef] [PubMed]
- Rendueles, O.; Ghigo, J.-M. Mechanisms of Competition in Biofilm Communities. Microbiol. Spectr. 2015, 3, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, N.M.; Martinez-Garcia, E.; Xavier, J.; Durham, W.M.; Kolter, R.; Kim, W.; Foster, K.R. Biofilm Formation As a Response to Ecological Competition. PLoS Biol. 2015, 13, e1002232. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef]
- Schluter, J.; Nadell, C.D.; Bassler, B.L.; Foster, K.R. Adhesion as a Weapon in Microbial Competition. ISME J. 2015, 9, 139–149. [Google Scholar] [CrossRef]
- Chassaing, B.; Cascales, E. Antibacterial Weapons: Targeted Destruction in the Microbiota. Trends Microbiol. 2018, 26, 329–338. [Google Scholar] [CrossRef]
- Platt, T.G.; Bever, J.D. Kin Competition and the Evolution of Cooperation. Trends Ecol. Evol. 2009, 24, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.J. Soft Selection, Hard Selection, Kin Selection, and Group Selection. Am. Nat. 1985, 125, 61–73. [Google Scholar] [CrossRef]
- Popat, R.; Crusz, S.A.; Messina, M.; Williams, P.; West, S.A.; Diggle, S.P. Quorum-Sensing and Cheating in Bacterial Biofilms. Proc. Biol. Sci. 2012, 279, 4765–4771. [Google Scholar] [CrossRef] [PubMed]
- Drescher, K.; Nadell, C.D.; Stone, H.A.; Wingreen, N.S.; Bassler, B.L. Solutions to the Public Goods Dilemma in Bacterial Biofilms. Curr. Biol. 2014, 24, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial Siderophores in Community and Host Interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef]
- Webb, J.S.; Thompson, L.S.; James, S.; Charlton, T.; Tolker-Nielsen, T.; Koch, B.; Givskov, M.; Kjelleberg, S. Cell Death in Pseudomonas aeruginosa Biofilm Development. J. Bacteriol. 2003, 185, 4585–4592. [Google Scholar] [CrossRef]
- Darch, S.E.; West, S.A.; Winzer, K.; Diggle, S.P. Density-Dependent Fitness Benefits in Quorum-Sensing Bacterial Populations. Proc. Natl. Acad. Sci. USA 2012, 109, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Van Gestel, J.; Vlamakis, H.; Kolter, R. Division of Labor in Biofilms: The Ecology of Cell Differentiation. Microbiol. Spectr. 2015, 3, MB-0002-2014. [Google Scholar] [CrossRef]
- Crabbé, A.; Jensen, P.Ø.; Bjarnsholt, T.; Coenye, T. Antimicrobial Tolerance and Metabolic Adaptations in Microbial Biofilms. Trends Microbiol. 2019, 27, 850–863. [Google Scholar] [CrossRef]
- Stewart, P.S.; Franklin, M.J. Physiological Heterogeneity in Biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef]
- Dragoš, A.; Kiesewalter, H.; Martin, M.; Hsu, C.-Y.; Hartmann, R.; Wechsler, T.; Eriksen, C.; Brix, S.; Drescher, K.; Stanley-Wall, N.; et al. Division of Labor during Biofilm Matrix Production. Curr. Biol. 2018, 28, 1903–1913.e5. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, B.; Murphy, C.D.; Casey, E. Comparison of Planktonic and Biofilm Cultures of Pseudomonas fluorescens DSM 8341 Cells Grown on Fluoroacetate. Appl. Environ. Microbiol. 2009, 75, 2899–2907. [Google Scholar] [CrossRef]
- Williamson, K.S.; Richards, L.A.; Perez-Osorio, A.C.; Pitts, B.; McInnerney, K.; Stewart, P.S.; Franklin, M.J. Heterogeneity in Pseudomonas aeruginosa Biofilms Includes Expression of Ribosome Hibernation Factors in the Antibiotic-Tolerant Subpopulation and Hypoxia-Induced Stress Response in the Metabolically Active Population. J. Bacteriol. 2012, 194, 2062–2073. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Composition and Metabolic Activities of Bacterial Biofilms Colonizing Food Residues in the Human Gut. Appl. Environ. Microbiol. 2006, 72, 6204–6211. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Alhede, M.; Alhede, M.; Eickhardt-Sørensen, S.R.; Moser, C.; Kühl, M.; Jensen, P.Ø.; Høiby, N. The in vivo Biofilm. Trends Microbiol. 2013, 21, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Folsom, J.P.; Richards, L.; Pitts, B.; Roe, F.; Ehrlich, G.D.; Parker, A.; Mazurie, A.; Stewart, P.S. Physiology of Pseudomonas aeruginosa in Biofilms as Revealed by Transcriptome Analysis. BMC Microbiol. 2010, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Sønderholm, M.; Kragh, K.N.; Koren, K.; Jakobsen, T.H.; Darch, S.E.; Alhede, M.; Jensen, P.Ø.; Whiteley, M.; Kühl, M.; Bjarnsholt, T. Pseudomonas aeruginosa Aggregate Formation in an Alginate Bead Model System Exhibits In Vivo -Like Characteristics. Appl. Environ. Microbiol. 2017, 83, e00113-17. [Google Scholar] [CrossRef]
- Cornforth, D.M.; Dees, J.L.; Ibberson, C.B.; Huse, H.K.; Mathiesen, I.H.; Kirketerp-Møller, K.; Wolcott, R.D.; Rumbaugh, K.P.; Bjarnsholt, T.; Whiteley, M. Pseudomonas aeruginosa Transcriptome during Human Infection. Proc. Natl. Acad. Sci. USA 2018, 115, E5125–E5134. [Google Scholar] [CrossRef]
- Rossi, E.; Falcone, M.; Molin, S.; Johansen, H.K. High-Resolution In Situ Transcriptomics of Pseudomonas aeruginosa Unveils Genotype Independent Patho-Phenotypes in Cystic Fibrosis Lungs. Nat. Commun. 2018, 9, 3459. [Google Scholar] [CrossRef]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef]
- Barken, K.B.; Pamp, S.J.; Yang, L.; Gjermansen, M.; Bertrand, J.J.; Klausen, M.; Givskov, M.; Whitchurch, C.B.; Engel, J.N.; Tolker-Nielsen, T. Roles of Type IV Pili, Flagellum-Mediated Motility and Extracellular DNA in the Formation of Mature Multicellular Structures in Pseudomonas aeruginosa Biofilms. Environ. Microbiol. 2008, 10, 2331–2343. [Google Scholar] [CrossRef]
- Inhülsen, S.; Aguilar, C.; Schmid, N.; Suppiger, A.; Riedel, K.; Eberl, L. Identification of Functions Linking Quorum Sensing with Biofilm Formation in Burkholderia cenocepacia H111. Microbiologyopen 2012, 1, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Biofilm Development. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Karatan, E.; Watnick, P. Signals, Regulatory Networks, and Materials That Build and Break Bacterial Biofilms. Microbiol. Mol. Biol. Rev. 2009, 73, 310–347. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, K.K. What Drives Bacteria to Produce a Biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef]
- Gambino, M.; Cappitelli, F. Mini-Review: Biofilm Responses to Oxidative Stress. Biofouling 2016, 32, 167–178. [Google Scholar] [CrossRef]
- Kang, D.; Kirienko, N.V. Interdependence between Iron Acquisition and Biofilm Formation in Pseudomonas aeruginosa. J. Microbiol. 2018, 56, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B. Antibiotic-Induced Biofilm Formation. Int. J. Artif. Organs. 2011, 34, 737–751. [Google Scholar] [CrossRef]
- Chodur, D.M.; Coulter, P.; Isaacs, J.; Pu, M.; Fernandez, N.; Waters, C.M.; Rowe-Magnus, D.A. Environmental Calcium Initiates a Feed-Forward Signaling Circuit That Regulates Biofilm Formation and Rugosity in Vibrio Vulnificus. mBio 2018, 9, e01377-18. [Google Scholar] [CrossRef]
- Hung, D.T.; Zhu, J.; Sturtevant, D.; Mekalanos, J.J. Bile Acids Stimulate Biofilm Formation in Vibrio Cholerae. Mol. Microbiol. 2006, 59, 193–201. [Google Scholar] [CrossRef]
- Koestler, B.J.; Waters, C.M. Intestinal GPS: Bile and Bicarbonate Control Cyclic Di-GMP to Provide Vibrio Cholerae Spatial Cues within the Small Intestine. Gut Microbes 2014, 5, 775–780. [Google Scholar] [CrossRef]
- Köseoğlu, V.K.; Hall, C.P.; Rodríguez-López, E.M.; Agaisse, H. The Autotransporter IcsA Promotes Shigella flexneri Biofilm Formation in the Presence of Bile Salts. Infect. Immun. 2019, 87, e00861-18. [Google Scholar] [CrossRef]
- McKenney, P.T.; Yan, J.; Vaubourgeix, J.; Becattini, S.; Lampen, N.; Motzer, A.; Larson, P.J.; Dannaoui, D.; Fujisawa, S.; Xavier, J.B.; et al. Intestinal Bile Acids Induce a Morphotype Switch in Vancomycin-Resistant Enterococcus That Facilitates Intestinal Colonization. Cell Host Microbe 2019, 25, 695–705.e5. [Google Scholar] [CrossRef]
- Pumbwe, L.; Skilbeck, C.A.; Nakano, V.; Avila-Campos, M.J.; Piazza, R.M.F.; Wexler, H.M. Bile Salts Enhance Bacterial Co-Aggregation, Bacterial-Intestinal Epithelial Cell Adhesion, Biofilm Formation and Antimicrobial Resistance of Bacteroides Fragilis. Microb. Pathog. 2007, 43, 78–87. [Google Scholar] [CrossRef]
- Kelly, S.M.; Lanigan, N.; O’Neill, I.J.; Bottacini, F.; Lugli, G.A.; Viappiani, A.; Turroni, F.; Ventura, M.; van Sinderen, D. Bifidobacterial Biofilm Formation Is a Multifactorial Adaptive Phenomenon in Response to Bile Exposure. Sci. Rep. 2020, 10, 11598. [Google Scholar] [CrossRef]
- López, M.; Blasco, L.; Gato, E.; Perez, A.; Fernández-Garcia, L.; Martínez-Martinez, L.; Fernández-Cuenca, F.; Rodríguez-Baño, J.; Pascual, A.; Bou, G.; et al. Response to Bile Salts in Clinical Strains of Acinetobacter baumannii Lacking the AdeABC Efflux Pump: Virulence Associated with Quorum Sensing. Front. Cell Infect. Microbiol. 2017, 7, 143. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Ling, N.; Shen, Y.; Zhang, D.; Liu, D.; Ou, D.; Wu, Q.; Ye, Y. Roles of TolC on Tolerance to Bile Salts and Biofilm Formation in Cronobacter malonaticus. J. Dairy Sci. 2021, 104, 9521–9531. [Google Scholar] [CrossRef]
- Ambalam, P.; Kondepudi, K.K.; Nilsson, I.; Wadström, T.; Ljungh, A. Bile Enhances Cell Surface Hydrophobicity and Biofilm Formation of Bifidobacteria. Appl. Biochem. Biotechnol. 2014, 172, 1970–1981. [Google Scholar] [CrossRef]
- Bechon, N.; Mihajlovic, J.; Lopes, A.-A.; Vendrell-Fernández, S.; Deschamps, J.; Briandet, R.; Sismeiro, O.; Martin-Verstraete, I.; Dupuy, B.; Ghigo, J.-M. Bacteroides thetaiotaomicron Uses a Widespread Extracellular DNase to Promote Bile-Dependent Biofilm Formation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bollinger, R.R.; Everett, M.L.; Palestrant, D.; Love, S.D.; Lin, S.S.; Parker, W. Human Secretory Immunoglobulin A May Contribute to Biofilm Formation in the Gut. Immunology 2003, 109, 580–587. [Google Scholar] [CrossRef]
- Everett, M.L.; Palestrant, D.; Miller, S.E.; Bollinger, R.R.; Parker, W. Immune Exclusion and Immune Inclusion: A New Model of Host-Bacterial Interactions in the Gut. Clin. Appl. Immunol. Rev. 2004, 4, 321–332. [Google Scholar] [CrossRef]
- Popowska, M.; Krawczyk-Balska, A.; Ostrowski, R.; Desvaux, M. InlL from Listeria monocytogenes Is Involved in Biofilm Formation and Adhesion to Mucin. Front. Microbiol. 2017, 8, 660. [Google Scholar] [CrossRef]
- Tu, Q.V.; McGuckin, M.A.; Mendz, G.L. Campylobacter jejuni Response to Human Mucin MUC2: Modulation of Colonization and Pathogenicity Determinants. J. Med. Microbiol. 2008, 57, 795–802. [Google Scholar] [CrossRef]
- Dwivedi, R.; Nothaft, H.; Garber, J.; Xin Kin, L.; Stahl, M.; Flint, A.; van Vliet, A.H.M.; Stintzi, A.; Szymanski, C.M. L-Fucose Influences Chemotaxis and Biofilm Formation in Campylobacter jejuni. Mol. Microbiol. 2016, 101, 575–589. [Google Scholar] [CrossRef]
- Motta, J.-P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; Da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen Sulfide Protects from Colitis and Restores Intestinal Microbiota Biofilm and Mucus Production. Inflamm. Bowel. Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef]
- Feraco, D.; Blaha, M.; Khan, S.; Green, J.M.; Plotkin, B.J. Host Environmental Signals and Effects on Biofilm Formation. Microb. Pathog. 2016, 99, 253–263. [Google Scholar] [CrossRef]
- Sperandio, V.; Torres, A.G.; Jarvis, B.; Nataro, J.P.; Kaper, J.B. Bacteria-Host Communication: The Language of Hormones. Proc. Natl. Acad. Sci. USA 2003, 100, 8951–8956. [Google Scholar] [CrossRef]
- Plotkin, B.J.; Wu, Z.; Ward, K.; Nadella, S.; Green, J.M.; Rumnani, B. Effect of Human Insulin on the Formation of Catheter-Associated E. coli biofilms. OJU 2014, 4, 49–56. [Google Scholar] [CrossRef]
- Klosowska, K.; Plotkin, B.J. Human Insulin Modulation of Escherichia coli Adherence and Chemotaxis. Am. J. Infect. Dis. 2006, 2, 197–200. [Google Scholar] [CrossRef]
- Scardaci, R.; Varese, F.; Manfredi, M.; Marengo, E.; Mazzoli, R.; Pessione, E. Enterococcus faecium NCIMB10415 Responds to Norepinephrine by Altering Protein Profiles and Phenotypic Characters. J. Proteom. 2021, 231, 104003. [Google Scholar] [CrossRef]
- Cambronel, M.; Nilly, F.; Mesguida, O.; Boukerb, A.M.; Racine, P.-J.; Baccouri, O.; Borrel, V.; Martel, J.; Fécamp, F.; Knowlton, R.; et al. Influence of Catecholamines (Epinephrine/Norepinephrine) on Biofilm Formation and Adhesion in Pathogenic and Probiotic Strains of Enterococcus faecalis. Front. Microbiol. 2020, 11, 1501. [Google Scholar] [CrossRef]
- Hiller, C.C.; Lucca, V.; Carvalho, D.; Borsoi, A.; Borges, K.A.; Furian, T.Q.; do Nascimento, V.P. Influence of Catecholamines on Biofilm Formation by Salmonella enteritidis. Microb. Pathog. 2019, 130, 54–58. [Google Scholar] [CrossRef]
- Maestre, J.R.; Aguilar, L.; Mateo, M.; Giménez, M.-J.; Méndez, M.-L.; Alou, L.; Granizo, J.-J.; Prieto, J. In vitro Interference of Tigecycline at Subinhibitory Concentrations on Biofilm Development by Enterococcus faecalis. J. Antimicrob. Chemother. 2012, 67, 1155–1158. [Google Scholar] [CrossRef]
- Ozma, M.A.; Khodadadi, E.; Rezaee, M.A.; Kamounah, F.S.; Asgharzadeh, M.; Ganbarov, K.; Aghazadeh, M.; Yousefi, M.; Pirzadeh, T.; Kafil, H.S. Induction of Proteome Changes Involved in Biofilm Formation of Enterococcus faecalis in Response to Gentamicin. Microb. Pathog. 2021, 157, 105003. [Google Scholar] [CrossRef]
- Yu, W.; Hallinen, K.M.; Wood, K.B. Interplay between Antibiotic Efficacy and Drug-Induced Lysis Underlies Enhanced Biofilm Formation at Subinhibitory Drug Concentrations. Antimicrob. Agents Chemother. 2018, 62, e01603-17. [Google Scholar] [CrossRef]
- Hoffman, L.R.; D’Argenio, D.A.; MacCoss, M.J.; Zhang, Z.; Jones, R.A.; Miller, S.I. Aminoglycoside Antibiotics Induce Bacterial Biofilm Formation. Nature 2005, 436, 1171–1175. [Google Scholar] [CrossRef]
- Sun, F.; Yuan, Q.; Wang, Y.; Cheng, L.; Li, X.; Feng, W.; Xia, P. Sub-Minimum Inhibitory Concentration Ceftazidime Inhibits Escherichia coli Biofilm Formation by Influencing the Levels of the IbpA Gene and Extracellular Indole. J. Chemother. 2020, 32, 7–14. [Google Scholar] [CrossRef]
- Alav, I.; Sutton, J.M.; Rahman, K.M. Role of Bacterial Efflux Pumps in Biofilm Formation. J. Antimicrob. Chemother. 2018, 73, 2003–2020. [Google Scholar] [CrossRef] [PubMed]
- Schembri, M.A.; Kjaergaard, K.; Klemm, P. Global Gene Expression in Escherichia coli Biofilms. Mol. Microbiol. 2003, 48, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. Regulation of Uptake and Processing of the Quorum-Sensing Autoinducer AI-2 in Escherichia coli. J. Bacteriol. 2005, 187, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Bay, D.C.; Stremick, C.A.; Slipski, C.J.; Turner, R.J. Secondary Multidrug Efflux Pump Mutants Alter Escherichia coli Biofilm Growth in the Presence of Cationic Antimicrobial Compounds. Res. Microbiol. 2017, 168, 208–221. [Google Scholar] [CrossRef]
- Matsumura, K.; Furukawa, S.; Ogihara, H.; Morinaga, Y. Roles of Multidrug Efflux Pumps on the Biofilm Formation of Escherichia coli K-12. Biocontrol. Sci. 2011, 16, 69–72. [Google Scholar] [CrossRef]
- Baugh, S.; Ekanayaka, A.S.; Piddock, L.J.V.; Webber, M.A. Loss of or Inhibition of All Multidrug Resistance Efflux Pumps of Salmonella enterica Serovar Typhimurium Results in Impaired Ability to Form a Biofilm. J. Antimicrob. Chemother. 2012, 67, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Teteneva, N.A.; Mart’yanov, S.V.; Esteban-López, M.; Kahnt, J.; Glatter, T.; Netrusov, A.I.; Plakunov, V.K.; Sourjik, V. Multiple Drug-Induced Stress Responses Inhibit Formation of Escherichia coli Biofilms. Appl. Environ. Microbiol. 2020, 86, e01113-20. [Google Scholar] [CrossRef]
- LeBlanc, J.G.; Chain, F.; Martín, R.; Bermúdez-Humarán, L.G.; Courau, S.; Langella, P. Beneficial Effects on Host Energy Metabolism of Short-Chain Fatty Acids and Vitamins Produced by Commensal and Probiotic Bacteria. Microb. Cell Fact. 2017, 16, 79. [Google Scholar] [CrossRef]
- Jacobson, A.; Lam, L.; Rajendram, M.; Tamburini, F.; Honeycutt, J.; Pham, T.; Van Treuren, W.; Pruss, K.; Stabler, S.R.; Lugo, K.; et al. A Gut Commensal-Produced Metabolite Mediates Colonization Resistance to Salmonella Infection. Cell Host Microbe 2018, 24, 296–307.e7. [Google Scholar] [CrossRef]
- Suzuki, I.; Shimizu, T.; Senpuku, H. Role of SCFAs for Fimbrillin-Dependent Biofilm Formation of Actinomyces oris. Microorganisms 2018, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Kawarai, T.; Narisawa, N.; Tuna, E.B.; Sato, N.; Tsugane, T.; Saeki, Y.; Ochiai, K.; Senpuku, H. Effects of Short-Chain Fatty Acids on Actinomyces naeslundii Biofilm Formation. Mol. Oral. Microbiol. 2013, 28, 354–365. [Google Scholar] [CrossRef]
- Lamas, A.; Regal, P.; Vázquez, B.; Cepeda, A.; Franco, C.M. Short Chain Fatty Acids Commonly Produced by Gut Microbiota Influence Salmonella enterica Motility, Biofilm Formation, and Gene Expression. Antibiotics 2019, 8, 265. [Google Scholar] [CrossRef]
- Hu, M.; Zhang, C.; Mu, Y.; Shen, Q.; Feng, Y. Indole Affects Biofilm Formation in Bacteria. Indian J. Microbiol. 2010, 50, 362–368. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, J. Indole as an Intercellular Signal in Microbial Communities. FEMS Microbiol. Rev. 2010, 34, 426–444. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.S.; Beyhan, S.; Saini, S.G.; Yildiz, F.H.; Bartlett, D.H. Indole Acts as an Extracellular Cue Regulating Gene Expression in Vibrio cholerae. J. Bacteriol. 2009, 191, 3504–3516. [Google Scholar] [CrossRef]
- Martino, P.D.; Fursy, R.; Bret, L.; Sundararaju, B.; Phillips, R.S. Indole Can Act as an Extracellular Signal to Regulate Biofilm Formation of Escherichia coli and Other Indole-Producing Bacteria. Can. J. Microbiol. 2003, 49, 443–449. [Google Scholar] [CrossRef]
- Sasaki-Imamura, T.; Yano, A.; Yoshida, Y. Production of Indole from L-Tryptophan and Effects of These Compounds on Biofilm Formation by Fusobacterium nucleatum ATCC 25586. Appl. Environ. Microbiol. 2010, 76, 4260–4268. [Google Scholar] [CrossRef] [PubMed]
- Rattanaphan, P.; Mittraparp-Arthorn, P.; Srinoun, K.; Vuddhakul, V.; Tansila, N. Indole Signaling Decreases Biofilm Formation and Related Virulence of Listeria monocytogenes. FEMS Microbiol. Lett. 2020, 367, fnaa116. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.D.S.; Hill, C. Gut Bacteriophage: Current Understanding and Challenges. Front. Endocrinol. 2019, 10, 784. [Google Scholar] [CrossRef]
- Lacqua, A.; Wanner, O.; Colangelo, T.; Martinotti, M.G.; Landini, P. Emergence of Biofilm-Forming Subpopulations upon Exposure of Escherichia coli to Environmental Bacteriophages. Appl. Environ. Microbiol. 2006, 72, 956–959. [Google Scholar] [CrossRef]
- Hosseinidoust, Z.; Tufenkji, N.; van de Ven, T.G.M. Formation of Biofilms under Phage Predation: Considerations Concerning a Biofilm Increase. Biofouling 2013, 29, 457–468. [Google Scholar] [CrossRef]
- Rossmann, F.S.; Racek, T.; Wobser, D.; Puchalka, J.; Rabener, E.M.; Reiger, M.; Hendrickx, A.P.A.; Diederich, A.-K.; Jung, K.; Klein, C.; et al. Phage-Mediated Dispersal of Biofilm and Distribution of Bacterial Virulence Genes Is Induced by Quorum Sensing. PLoS Pathog. 2015, 11, e1004653. [Google Scholar] [CrossRef] [PubMed]
- García-Contreras, R.; Zhang, X.-S.; Kim, Y.; Wood, T.K. Protein Translation and Cell Death: The Role of Rare TRNAs in Biofilm Formation and in Activating Dormant Phage Killer Genes. PLoS ONE 2008, 3, e2394. [Google Scholar] [CrossRef] [PubMed]
- Papenfort, K.; Silpe, J.E.; Schramma, K.R.; Cong, J.-P.; Seyedsayamdost, M.R.; Bassler, B.L. A Vibrio cholerae Autoinducer–Receptor Pair That Controls Biofilm Formation. Nat. Chem. Biol. 2017, 13, 551–557. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A Host-Produced Quorum-Sensing Autoinducer Controls a Phage Lysis-Lysogeny Decision. Cell 2019, 176, 268–280.e13. [Google Scholar] [CrossRef]
- Fu, Y.; Luo, Y.; Grinspan, A.M. Epidemiology of Community-Acquired and Recurrent Clostridioides difficile Infection. Ther. Adv. Gastroenterol. 2021, 14, 17562848211016248. [Google Scholar] [CrossRef] [PubMed]
- Dubberke, E.R.; Olsen, M.A. Burden of Clostridium difficile on the Healthcare System. Clin. Infect. Dis. 2012, 55 (Suppl. S2), S88–S92. [Google Scholar] [CrossRef] [PubMed]
- Reveles, K.R.; Lawson, K.A.; Mortensen, E.M.; Pugh, M.J.V.; Koeller, J.M.; Argamany, J.R.; Frei, C.R. National Epidemiology of Initial and Recurrent Clostridium difficile Infection in the Veterans Health Administration from 2003 to 2014. PLoS ONE 2017, 12, e0189227. [Google Scholar] [CrossRef]
- Jon, J.V.; Mark, H.W.; Jane, F. Antimicrobial Resistance Progression in the United Kingdom: A Temporal Comparison of Clostridioides difficile Antimicrobial Susceptibilities. Anaerobe 2021, 70, 102385. [Google Scholar] [CrossRef]
- Czepiel, J.; Dróżdż, M.; Pituch, H.; Kuijper, E.J.; Perucki, W.; Mielimonka, A.; Goldman, S.; Wultańska, D.; Garlicki, A.; Biesiada, G. Clostridium difficile Infection: Review. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1211–1221. [Google Scholar] [CrossRef]
- Simor, A.E. Diagnosis, Management, and Prevention of Clostridium difficile Infection in Long-Term Care Facilities: A Review. J. Am. Geriatr. Soc. 2010, 58, 1556–1564. [Google Scholar] [CrossRef]
- Lawley, T.D.; Clare, S.; Walker, A.W.; Goulding, D.; Stabler, R.A.; Croucher, N.; Mastroeni, P.; Scott, P.; Raisen, C.; Mottram, L.; et al. Antibiotic Treatment of Clostridium difficile Carrier Mice Triggers a Supershedder State, Spore-Mediated Transmission, and Severe Disease in Immunocompromised Hosts. Infect. Immun. 2009, 77, 3661–3669. [Google Scholar] [CrossRef] [PubMed]
- Pruss, K.M.; Sonnenburg, J.L.C. Difficile Exploits a Host Metabolite Produced during Toxin-Mediated Disease. Nature 2021, 593, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Abt, M.C.; McKenney, P.T.; Pamer, E.G. Clostridium difficile Colitis: Pathogenesis and Host Defence. Nat. Rev. Microbiol. 2016, 14, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.; Andersson, F.L.; Madin-Warburton, M. Burden of Clostridioides difficile Infection (CDI)—A Systematic Review of the Epidemiology of Primary and Recurrent CDI. BMC Infect. Dis. 2021, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barbut, F.; Richard, A.; Hamadi, K.; Chomette, V.; Burghoffer, B.; Petit, J.-C. Epidemiology of Recurrences or Reinfections of Clostridium difficile-Associated Diarrhea. J. Clin. Microbiol. 2000, 38, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Deakin, L.J.; Clare, S.; Fagan, R.P.; Dawson, L.F.; Pickard, D.J.; West, M.R.; Wren, B.W.; Fairweather, N.F.; Dougan, G.; Lawley, T.D. The Clostridium difficile Spo0A Gene Is a Persistence and Transmission Factor. Infect. Immun. 2012, 80, 2704–2711. [Google Scholar] [CrossRef]
- Castro-Córdova, P.; Mora-Uribe, P.; Reyes-Ramírez, R.; Cofré-Araneda, G.; Orozco-Aguilar, J.; Brito-Silva, C.; Mendoza-León, M.J.; Kuehne, S.A.; Minton, N.P.; Pizarro-Guajardo, M.; et al. Entry of Spores into Intestinal Epithelial Cells Contributes to Recurrence of Clostridioides difficile Infection. Nat. Commun. 2021, 12, 1140. [Google Scholar] [CrossRef]
- Scherr, T.D.; Heim, C.E.; Morrison, J.M.; Kielian, T. Hiding in Plain Sight: Interplay between Staphylococcal Biofilms and Host Immunity. Front. Immunol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Thomsen, K.; Kobayashi, O.; Kishi, K.; Shirai, R.; Østrup Jensen, P.; Heydorn, A.; Hentzer, M.; Calum, H.; Christophersen, L.; Høiby, N.; et al. Animal Models of Chronic and Recurrent Pseudomonas aeruginosa Lung Infection: Significance of Macrolide Treatment. APMIS 2021. [Google Scholar] [CrossRef]
- Frost, L.R.; Cheng, J.K.J.; Unnikrishnan, M. Clostridioides difficile Biofilms: A Mechanism of Persistence in the Gut? PLoS Pathog. 2021, 17, e1009348. [Google Scholar] [CrossRef] [PubMed]
- Donelli, G.; Vuotto, C.; Cardines, R.; Mastrantonio, P. Biofilm-Growing Intestinal Anaerobic Bacteria. FEMS Immunol. Med. Microbiol. 2012, 65, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.F.; Valiente, E.; Faulds-Pain, A.; Donahue, E.H.; Wren, B.W. Characterisation of Clostridium difficile Biofilm Formation, a Role for Spo0A. PLoS ONE 2012, 7, e50527. [Google Scholar] [CrossRef]
- Dubois, T.; Tremblay, Y.D.N.; Hamiot, A.; Martin-Verstraete, I.; Deschamps, J.; Monot, M.; Briandet, R.; Dupuy, B. A Microbiota-Generated Bile Salt Induces Biofilm Formation in Clostridium difficile. NPJ Biofilms Microbiomes 2019, 5, 14. [Google Scholar] [CrossRef]
- Poquet, I.; Saujet, L.; Canette, A.; Monot, M.; Mihajlovic, J.; Ghigo, J.-M.; Soutourina, O.; Briandet, R.; Martin-Verstraete, I.; Dupuy, B. Clostridium difficile Biofilm: Remodeling Metabolism and Cell Surface to Build a Sparse and Heterogeneously Aggregated Architecture. Front. Microbiol. 2018, 9, 2084. [Google Scholar] [CrossRef]
- Ghigo, J.-M. Natural Conjugative Plasmids Induce Bacterial Biofilm Development. Nature 2001, 412, 4. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Chilton, C.H.; Todhunter, S.L.; Nicholson, S.; Freeman, J.; Baines, S.D.; Wilcox, M.H. Development and Validation of a Chemostat Gut Model to Study Both Planktonic and Biofilm Modes of Growth of Clostridium difficile and Human Microbiota. PLoS ONE 2014, 9, e88396. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.S.; Wilcox, M.H.; Chilton, C.H. An In vitro Model of the Human Colon: Studies of Intestinal Biofilms and Clostridium difficile Infection. Methods Mol. Biol. 2016, 1476, 223–234. [Google Scholar] [CrossRef]
- Brauer, M.; Lassek, C.; Hinze, C.; Hoyer, J.; Becher, D.; Jahn, D.; Sievers, S.; Riedel, K. What’s a Biofilm?—How the Choice of the Biofilm Model Impacts the Protein Inventory of Clostridioides difficile. Front. Microbiol. 2021, 12, 1453. [Google Scholar] [CrossRef]
- Semenyuk, E.G.; Laning, M.L.; Foley, J.; Johnston, P.F.; Knight, K.L.; Gerding, D.N.; Driks, A. Spore Formation and Toxin Production in Clostridium difficile Biofilms. PLoS ONE 2014, 9, e87757. [Google Scholar] [CrossRef]
- James, G.A.; Chesnel, L.; Boegli, L.; deLancey Pulcini, E.; Fisher, S.; Stewart, P.S. Analysis of Clostridium difficile Biofilms: Imaging and Antimicrobial Treatment. J. Antimicrob. Chemother. 2018, 73, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, Y.D.N.; Durand, B.A.R.; Hamiot, A.; Martin-Verstraete, I.; Oberkampf, M.; Monot, M.; Dupuy, B. Metabolic Adaption to Extracellular Pyruvate Triggers Biofilm Formation in Clostridioides difficile. ISME J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pantaléon, V.; Monot, M.; Eckert, C.; Hoys, S.; Collignon, A.; Janoir, C.; Candela, T. Clostridium difficile Forms Variable Biofilms on Abiotic Surface. Anaerobe 2018, 53, 34–37. [Google Scholar] [CrossRef]
- Semenyuk, E.G.; Poroyko, V.A.; Johnston, P.F.; Jones, S.E.; Knight, K.L.; Gerding, D.N.; Driks, A. Analysis of Bacterial Communities during Clostridium difficile Infection in the Mouse. Infect. Immun. 2015, 83, 4383–4391. [Google Scholar] [CrossRef]
- Pantaléon, V.; Soavelomandroso, A.P.; Bouttier, S.; Briandet, R.; Roxas, B.; Chu, M.; Collignon, A.; Janoir, C.; Vedantam, G.; Candela, T. The Clostridium difficile Protease Cwp84 Modulates Both Biofilm Formation and Cell-Surface Properties. PLoS ONE 2015, 10, e0124971. [Google Scholar] [CrossRef] [PubMed]
- Soavelomandroso, A.P.; Gaudin, F.; Hoys, S.; Nicolas, V.; Vedantam, G.; Janoir, C.; Bouttier, S. Biofilm Structures in a Mono-Associated Mouse Model of Clostridium difficile Infection. Front. Microbiol. 2017, 8, 2086. [Google Scholar] [CrossRef]
- Buckley, A.M.; Spencer, J.; Candlish, D.; Irvine, J.J.; Douce, G.R. Infection of Hamsters with the UK Clostridium difficile Ribotype 027 Outbreak Strain R20291. J. Med. Microbiol. 2011, 60, 1174–1180. [Google Scholar] [CrossRef]
- Rainha, K.; Fernandes Ferreira, R.; Trindade, C.N.R.; Carneiro, L.G.; Penna, B.; Endres, B.T.; Begum, K.; Alam, M.J.; Garey, K.W.; Domingues Regina Maria, C.P.; et al. Characterization of Clostridium difficile Ribotypes in Domestic Dogs in Rio de Janeiro, Brazil. Anaerobe 2019, 58, 22–29. [Google Scholar] [CrossRef]
- Martínez-Meléndez, A.; Morfin-Otero, R.; Villarreal-Treviño, L.; Baines, S.D.; Camacho-Ortíz, A.; Garza-González, E. Analysis of Biofilm Production and Expression of Adhesion Structures of Circulating Clostridium difficile Strains from Mexico. Enferm. Infecc. y Microbiol. Clínica 2021. [Google Scholar] [CrossRef] [PubMed]
- Barkin, J.A.; Sussman, D.A.; Fifadara, N.; Barkin, J.S. Clostridium difficile Infection and Patient-Specific Antimicrobial Resistance Testing Reveals a High Metronidazole Resistance Rate. Dig. Dis. Sci. 2017, 62, 1035–1042. [Google Scholar] [CrossRef]
- Ðapa, T.; Leuzzi, R.; Ng, Y.K.; Baban, S.T.; Adamo, R.; Kuehne, S.A.; Scarselli, M.; Minton, N.P.; Serruto, D.; Unnikrishnan, M. Multiple Factors Modulate Biofilm Formation by the Anaerobic Pathogen Clostridium difficile. J. Bacteriol. 2013, 195, 545–555. [Google Scholar] [CrossRef]
- Vuotto, C.; Moura, I.; Barbanti, F.; Donelli, G.; Spigaglia, P. Subinhibitory Concentrations of Metronidazole Increase Biofilm Formation in Clostridium difficile Strains. Pathog. Dis. 2016, 74, ftv114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Louie, T.J.; Gerding, D.N.; Cornely, O.A.; Chasan-Taber, S.; Fitts, D.; Gelone, S.P.; Broom, C.; Davidson, D.M. For the Polymer Alternative for CDI Treatment (PACT) investigators Vancomycin, Metronidazole, or Tolevamer for Clostridium difficile Infection: Results From Two Multinational, Randomized, Controlled Trials. Clin. Infect. Dis. 2014, 59, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Dawson, L.F.; Peltier, J.; Hall, C.L.; Harrison, M.A.; Derakhshan, M.; Shaw, H.A.; Fairweather, N.F.; Wren, B.W. Extracellular DNA, Cell Surface Proteins and c-Di-GMP Promote Biofilm Formation in Clostridioides difficile. Sci. Rep. 2021, 11, 3244. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.; Lowrance, B.; Anderson, A.C.; Weadge, J.T. Identification of the Clostridial Cellulose Synthase and Characterization of the Cognate Glycosyl Hydrolase, CcsZ. PLoS ONE 2020, 15, e0242686. [Google Scholar] [CrossRef]
- Dannheim, H.; Will, S.E.; Schomburg, D.; Neumann-Schaal, M. Clostridioides difficile 630Δerm in Silico and in vivo—Quantitative Growth and Extensive Polysaccharide Secretion. FEBS Open Bio. 2017, 7, 602–615. [Google Scholar] [CrossRef]
- Barnes, A.M.T.; Ballering, K.S.; Leibman, R.S.; Wells, C.L.; Dunny, G.M. Enterococcus faecalis Produces Abundant Extracellular Structures Containing DNA in the Absence of Cell Lysis during Early Biofilm Formation. mBio 2012, 3, e00193-12. [Google Scholar] [CrossRef]
- Slater, R.T.; Frost, L.R.; Jossi, S.E.; Millard, A.D.; Unnikrishnan, M. Clostridioides difficile LuxS Mediates Inter-Bacterial Interactions within Biofilms. Sci. Rep. 2019, 9, 9903. [Google Scholar] [CrossRef]
- Maikova, A.; Peltier, J.; Boudry, P.; Hajnsdorf, E.; Kint, N.; Monot, M.; Poquet, I.; Martin-Verstraete, I.; Dupuy, B.; Soutourina, O. Discovery of New Type I Toxin–Antitoxin Systems Adjacent to CRISPR Arrays in Clostridium difficile. Nucleic Acids Res. 2018, 46, 4733–4751. [Google Scholar] [CrossRef]
- El Meouche, I.; Peltier, J. Toxin Release Mediated by the Novel Autolysin Cwp19 in Clostridium difficile. Microb. Cell 2018, 5, 421–423. [Google Scholar] [CrossRef]
- Wydau-Dematteis, S.; El Meouche, I.; Courtin, P.; Hamiot, A.; Lai-Kuen, R.; Saubaméa, B.; Fenaille, F.; Butel, M.-J.; Pons, J.-L.; Dupuy, B.; et al. Cwp19 Is a Novel Lytic Transglycosylase Involved in Stationary-Phase Autolysis Resulting in Toxin Release in Clostridium difficile. mBio 2018, 9, e00648-18. [Google Scholar] [CrossRef]
- Maldarelli, G.A.; Piepenbrink, K.H.; Scott, A.J.; Freiberg, J.A.; Song, Y.; Achermann, Y.; Ernst, R.K.; Shirtliff, M.E.; Sundberg, E.J.; Donnenberg, M.S.; et al. Type IV Pili Promote Early Biofilm Formation by Clostridium difficile. Pathog. Dis. 2016, 74, ftw061. [Google Scholar] [CrossRef]
- McKee, R.W.; Aleksanyan, N.; Garrett, E.M.; Tamayo, R. Type IV Pili Promote Clostridium difficile Adherence and Persistence in a Mouse Model of Infection. Infect. Immun. 2018, 86, e00943-17. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, C.; Janoir, C.; Barc, M.-C.; Collignon, A.; Karjalainen, T. Identification and Characterization of a Fibronectin-Binding Protein from Clostridium difficile. Microbiology 2003, 149, 2779–2787. [Google Scholar] [CrossRef] [PubMed]
- Tulli, L.; Marchi, S.; Petracca, R.; Shaw, H.A.; Fairweather, N.F.; Scarselli, M.; Soriani, M.; Leuzzi, R. CbpA: A Novel Surface Exposed Adhesin of Clostridium difficile Targeting Human Collagen. Cell Microbiol. 2013, 15, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Valiente, E.; Bouché, L.; Hitchen, P.; Faulds-Pain, A.; Songane, M.; Dawson, L.F.; Donahue, E.; Stabler, R.A.; Panico, M.; Morris, H.R.; et al. Role of Glycosyltransferases Modifying Type B Flagellin of Emerging Hypervirulent Clostridium difficile Lineages and Their Impact on Motility and Biofilm Formation. J. Biol. Chem. 2016, 291, 25450–25461. [Google Scholar] [CrossRef]
- Faulds-Pain, A.; Twine, S.M.; Vinogradov, E.; Strong, P.C.R.; Dell, A.; Buckley, A.M.; Douce, G.R.; Valiente, E.; Logan, S.M.; Wren, B.W. The Post-Translational Modification of the Clostridium difficile Flagellin Affects Motility, Cell Surface Properties and Virulence. Mol. Microbiol. 2014, 94, 272–289. [Google Scholar] [CrossRef]
- Sievers, S.; Metzendorf, N.G.; Dittmann, S.; Troitzsch, D.; Gast, V.; Tröger, S.M.; Wolff, C.; Zühlke, D.; Hirschfeld, C.; Schlüter, R.; et al. Differential View on the Bile Acid Stress Response of Clostridioides difficile. Front. Microbiol. 2019, 10, 258. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Ewin, D.; Moura, I.B.; Wilcox, M.H.; Douce, G.R. Insights into the Regulatory Mechanisms of Clostridioides difficile Biofilm Formation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kearns, D.B.; Chu, F.; Branda, S.S.; Kolter, R.; Losick, R. A Master Regulator for Biofilm Formation by Bacillus subtilis. Mol. Microbiol. 2005, 55, 739–749. [Google Scholar] [CrossRef]
- Lewis, R.J.; Brannigan, J.A.; Offen, W.A.; Smith, I.; Wilkinson, A.J. An Evolutionary Link between Sporulation and Prophage Induction in the Structure of a Repressor:Anti-Repressor Complex11Edited by J. M. Thornton. J. Mol. Biol. 1998, 283, 907–912. [Google Scholar] [CrossRef]
- Girinathan, B.P.; Ou, J.; Dupuy, B.; Govind, R. Pleiotropic Roles of Clostridium difficile Sin Locus. PLoS Pathog. 2018, 14, e1006940. [Google Scholar] [CrossRef] [PubMed]
- Rezzonico, F.; Duffy, B. Lack of Genomic Evidence of AI-2 Receptors Suggests a Non-Quorum Sensing Role for LuxS in Most Bacteria. BMC Microbiol. 2008, 8, 154. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.J.; Clare, S.; Goulding, D.; Faulds-Pain, A.; Barquist, L.; Browne, H.P.; Pettit, L.; Dougan, G.; Lawley, T.D.; Wren, B.W. The Agr Locus Regulates Virulence and Colonization Genes in Clostridium difficile 027. J. Bacteriol. 2013, 195, 3672–3681. [Google Scholar] [CrossRef]
- Soutourina, O.A.; Monot, M.; Boudry, P.; Saujet, L.; Pichon, C.; Sismeiro, O.; Semenova, E.; Severinov, K.; Le Bouguenec, C.; Coppée, J.-Y.; et al. Genome-Wide Identification of Regulatory RNAs in the Human Pathogen Clostridium difficile. PLoS Genet. 2013, 9, e1003493. [Google Scholar] [CrossRef] [PubMed]
- McKee, R.W.; Harvest, C.K.; Tamayo, R. Cyclic Diguanylate Regulates Virulence Factor Genes via Multiple Riboswitches in Clostridium difficile. mSphere 2018, 3, e00423-18. [Google Scholar] [CrossRef]
- Arato, V.; Gasperini, G.; Giusti, F.; Ferlenghi, I.; Scarselli, M.; Leuzzi, R. Dual Role of the Colonization Factor CD2831 in Clostridium difficile Pathogenesis. Sci. Rep. 2019, 9, 5554. [Google Scholar] [CrossRef]
- Corver, J.; Cordo’, V.; van Leeuwen, H.C.; Klychnikov, O.I.; Hensbergen, P.J. Covalent Attachment and Pro-Pro Endopeptidase (PPEP-1)-Mediated Release of Clostridium difficile Cell Surface Proteins Involved in Adhesion. Mol. Microbiol. 2017, 105, 663–673. [Google Scholar] [CrossRef]
- Boudry, P.; Gracia, C.; Monot, M.; Caillet, J.; Saujet, L.; Hajnsdorf, E.; Dupuy, B.; Martin-Verstraete, I.; Soutourina, O. Pleiotropic Role of the RNA Chaperone Protein Hfq in the Human Pathogen Clostridium difficile. J. Bacteriol. 2014, 196, 3234–3248. [Google Scholar] [CrossRef]
- Cuenot, E.; Garcia-Garcia, T.; Douche, T.; Gorgette, O.; Courtin, P.; Denis-Quanquin, S.; Hoys, S.; Tremblay, Y.D.N.; Matondo, M.; Chapot-Chartier, M.-P.; et al. The Ser/Thr Kinase PrkC Participates in Cell Wall Homeostasis and Antimicrobial Resistance in Clostridium difficile. Infect. Immun. 2019, 87, e00005-19. [Google Scholar] [CrossRef]
- Jain, S.; Smyth, D.; O’Hagan, B.M.G.; Heap, J.T.; McMullan, G.; Minton, N.P.; Ternan, N.G. Inactivation of the DnaK Gene in Clostridium difficile 630 Δerm Yields a Temperature-Sensitive Phenotype and Increases Biofilm-Forming Ability. Sci. Rep. 2017, 7, 17522. [Google Scholar] [CrossRef] [PubMed]
- Walter, B.M.; Cartman, S.T.; Minton, N.P.; Butala, M.; Rupnik, M. The SOS Response Master Regulator LexA Is Associated with Sporulation, Motility and Biofilm Formation in Clostridium difficile. PLoS ONE 2015, 10, e0144763. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Ribis, J.W.; Garrett, E.M.; Trzilova, D.; Kim, A.; Sekulovic, O.; Mead, E.A.; Pak, T.; Zhu, S.; Deikus, G.; et al. Epigenomic Characterization of Clostridioides difficile Finds a Conserved DNA Methyltransferase That Mediates Sporulation and Pathogenesis. Nat. Microbiol. 2020, 5, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Trzilova, D.; Anjuwon-Foster, B.R.; Torres Rivera, D.; Tamayo, R. Rho Factor Mediates Flagellum and Toxin Phase Variation and Impacts Virulence in Clostridioides difficile. PLoS Pathog. 2020, 16, e1008708. [Google Scholar] [CrossRef] [PubMed]
- Garrett, E.M.; Sekulovic, O.; Wetzel, D.; Jones, J.B.; Edwards, A.N.; Vargas-Cuebas, G.; McBride, S.M.; Tamayo, R. Phase Variation of a Signal Transduction System Controls Clostridioides difficile Colony Morphology, Motility, and Virulence. PLoS Biol. 2019, 17, e3000379. [Google Scholar] [CrossRef] [PubMed]
- Sekulovic, O.; Mathias Garrett, E.; Bourgeois, J.; Tamayo, R.; Shen, A.; Camilli, A. Genome-Wide Detection of Conservative Site-Specific Recombination in Bacteria. PLoS Genet. 2018, 14, e1007332. [Google Scholar] [CrossRef]
- Emerson, J.E.; Reynolds, C.B.; Fagan, R.P.; Shaw, H.A.; Goulding, D.; Fairweather, N.F. A Novel Genetic Switch Controls Phase Variable Expression of CwpV, a Clostridium difficile Cell Wall Protein. Mol. Microbiol. 2009, 74, 541–556. [Google Scholar] [CrossRef]
- Anjuwon-Foster, B.R.; Tamayo, R. A Genetic Switch Controls the Production of Flagella and Toxins in Clostridium difficile. PLoS Genet. 2017, 13, e1006701. [Google Scholar] [CrossRef]
- Reyes Ruiz, L.M.; Williams, C.L.; Tamayo, R. Enhancing Bacterial Survival through Phenotypic Heterogeneity. PLoS Pathog. 2020, 16, e1008439. [Google Scholar] [CrossRef]
- Cargill, J.S.; Upton, M. Low Concentrations of Vancomycin Stimulate Biofilm Formation in Some Clinical Isolates of Staphylococcus epidermidis. J. Clin. Pathol. 2009, 62, 1112–1116. [Google Scholar] [CrossRef]
- Bedran, T.B.L.; Grignon, L.; Spolidorio, D.P.; Grenier, D. Subinhibitory Concentrations of Triclosan Promote Streptococcus Mutans Biofilm Formation and Adherence to Oral Epithelial Cells. PLoS ONE 2014, 9, e89059. [Google Scholar] [CrossRef]
- Wu, S.; Li, X.; Gunawardana, M.; Maguire, K.; Guerrero-Given, D.; Schaudinn, C.; Wang, C.; Baum, M.M.; Webster, P. Beta- Lactam Antibiotics Stimulate Biofilm Formation in Non-Typeable Haemophilus influenzae by Up-Regulating Carbohydrate Metabolism. PLoS ONE 2014, 9, e99204. [Google Scholar] [CrossRef]
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 2018, 66, e1–e48. [Google Scholar] [CrossRef]
- Wultańska, D.; Piotrowski, M.; Pituch, H. The Effect of Berberine Chloride and/or Its Combination with Vancomycin on the Growth, Biofilm Formation, and Motility of Clostridioides difficile. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1391–1399. [Google Scholar] [CrossRef]
- Vardakas, K.Z.; Polyzos, K.A.; Patouni, K.; Rafailidis, P.I.; Samonis, G.; Falagas, M.E. Treatment Failure and Recurrence of Clostridium difficile Infection Following Treatment with Vancomycin or Metronidazole: A Systematic Review of the Evidence. Int. J. Antimicrob. Agents. 2012, 40, 1–8. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.E.; Williams, K.B.; Whitehead, T.R.; Heuman, D.M.; Hylemon, P.B. Development and Application of a Polymerase Chain Reaction Assay for the Detection and Enumeration of Bile Acid 7α-Dehydroxylating Bacteria in Human Feces. Clin. Chim. Acta 2003, 331, 127–134. [Google Scholar] [CrossRef]
- Piotrowski, M.; Wultańska, D.; Obuch-Woszczatyński, P.; Pituch, H. Fructooligosaccharides and Mannose Affect Clostridium difficile Adhesion and Biofilm Formation in a Concentration-Dependent Manner. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 1975–1984. [Google Scholar] [CrossRef]
- Pereira, F.C.; Wasmund, K.; Cobankovic, I.; Jehmlich, N.; Herbold, C.W.; Lee, K.S.; Sziranyi, B.; Vesely, C.; Decker, T.; Stocker, R.; et al. Rational Design of a Microbial Consortium of Mucosal Sugar Utilizers Reduces Clostridium difficile Colonization. Nat. Commun. 2020, 11, 5104. [Google Scholar] [CrossRef]
- Engevik, M.A.; Engevik, A.C.; Engevik, K.A.; Auchtung, J.M.; Chang-Graham, A.L.; Ruan, W.; Luna, R.A.; Hyser, J.M.; Spinler, J.K.; Versalovic, J. Mucin-Degrading Microbes Release Monosaccharides That Chemoattract Clostridioides difficile and Facilitate Colonization of the Human Intestinal Mucus Layer. ACS Infect. Dis. 2020, 7, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision Microbiome Restoration of Bile Acid-Mediated Resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef]
- Normington, C.; Moura, I.B.; Bryant, J.A.; Ewin, D.J.; Clark, E.V.; Kettle, M.J.; Harris, H.C.; Spittal, W.; Davis, G.; Henn, M.R.; et al. Biofilms Harbour Clostridioides difficile, Serving as a Reservoir for Recurrent Infection. NPJ Biofilms Microbiomes 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial Biofilm and Associated Infections. J. Chin. Med. Assoc. 2018, 81, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.B.; Wright, J.R.; Fowler, M.; McLimans, C.J.; Tokarev, V.; Amaniera, I.; Baker, O.; Wong, H.-T.; Brabec, J.; Drucker, R.; et al. Integrated Meta-Omics Reveals a Fungus-Associated Bacteriome and Distinct Functional Pathways in Clostridioides difficile Infection. mSphere 2019, 4, e00454-19. [Google Scholar] [CrossRef] [PubMed]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial Biofilms: From the Natural Environment to Infectious Diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Sauer, K.; Camper, A.K.; Ehrlich, G.D.; Costerton, J.W.; Davies, D.G. Pseudomonas aeruginosa Displays Multiple Phenotypes during Development as a Biofilm. J. Bacteriol. 2002, 184, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Yu, Y.; Gozzi, K.; Chen, Y.; Guo, J.; Chai, Y. Genome-Wide Investigation of Biofilm Formation in Bacillus cereus. Appl. Environ. Microbiol. 2017, 83, e00561-17. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Stoodley, P. Evolving Concepts in Biofilm Infections. Cell Microbiol. 2009, 11, 1034–1043. [Google Scholar] [CrossRef]
- Mathur, H.; Rea, M.C.; Cotter, P.D.; Hill, C.; Ross, R.P. The Efficacy of Thuricin CD, Tigecycline, Vancomycin, Teicoplanin, Rifampicin and Nitazoxanide, Independently and in Paired Combinations against Clostridium difficile Biofilms and Planktonic Cells. Gut Pathog. 2016, 8, 20. [Google Scholar] [CrossRef]
- Mah, T.-F.C.; O’Toole, G.A. Mechanisms of Biofilm Resistance to Antimicrobial Agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Jefferson, K.K.; Goldmann, D.A.; Pier, G.B. Use of Confocal Microscopy to Analyze the Rate of Vancomycin Penetration through Staphylococcus aureus Biofilms. Antimicrob. Agents Chemother. 2005, 49, 2467–2473. [Google Scholar] [CrossRef]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of Antibiotic Penetration, Oxygen Limitation, and Low Metabolic Activity to Tolerance of Pseudomonas aeruginosa Biofilms to Ciprofloxacin and Tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef]
- Bagge, N.; Schuster, M.; Hentzer, M.; Ciofu, O.; Givskov, M.; Greenberg, E.P.; Høiby, N. Pseudomonas aeruginosa Biofilms Exposed to Imipenem Exhibit Changes in Global Gene Expression and β-Lactamase and Alginate Production. Antimicrob. Agents Chemother. 2004, 48, 1175–1187. [Google Scholar] [CrossRef]
- Bakkeren, E.; Huisman, J.S.; Fattinger, S.A.; Hausmann, A.; Furter, M.; Egli, A.; Slack, E.; Sellin, M.E.; Bonhoeffer, S.; Regoes, R.R.; et al. Salmonella Persisters Promote the Spread of Antibiotic Resistance Plasmids in the Gut. Nature 2019, 573, 276–280. [Google Scholar] [CrossRef]
- Álvarez, R.; Inostroza, O.; Garavaglia, M.; Minton, N.P.; Paredes-Sabja, D.; Gil, F. Effect of Antibiotic Treatment on the Formation of Non-Spore Clostridium difficile Persister-like Cells. J. Antimicrob. Chemother. 2018, 73, 2396–2399. [Google Scholar] [CrossRef]
- Pettit, L.J.; Browne, H.P.; Yu, L.; Smits, W.; Fagan, R.P.; Barquist, L.; Martin, M.J.; Goulding, D.; Duncan, S.H.; Flint, H.J.; et al. Functional Genomics Reveals That Clostridium difficile Spo0A Coordinates Sporulation, Virulence and Metabolism. BMC Genom. 2014, 15, 160. [Google Scholar] [CrossRef]
- Clevers, H. The Intestinal Crypt, A Prototype Stem Cell Compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Sjövall, H.; Hansson, G.C. The Gastrointestinal Mucus System in Health and Disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Hansson, G.C. Immunological Aspects of Intestinal Mucus and Mucins. Nat. Rev. Immunol. 2016, 16, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Buckley, A.M.; Moura, I.B.; Arai, N.; Spittal, W.; Clark, E.; Nishida, Y.; Harris, H.C.; Bentley, K.; Davis, G.; Wang, D.; et al. Trehalose-Induced Remodelling of the Human Microbiota Affects Clostridioides difficile Infection Outcome in an In vitro Colonic Model: A Pilot Study. Front. Cell. Infect. Microbiol. 2021, 11, 670935. [Google Scholar] [CrossRef]
- Abbas, A.; Zackular, J.P. Microbe–Microbe Interactions during Clostridioides difficile Infection. Curr. Opin. Microbiol. 2020, 53, 19–25. [Google Scholar] [CrossRef]
- Jenior, M.L.; Leslie, J.L.; Powers, D.A.; Garrett, E.M.; Walker, K.A.; Dickenson, M.E.; Petri, W.A.; Tamayo, R.; Papin, J.A. Conserved Virulence-Linked Metabolic Reprogramming in Clostridioides difficile Identified through Genome-Scale Metabolic Network Analysis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Henson, M.A. Computational modeling of the gut microbiota reveals putative metabolic mechanisms of recurrent Clostridioides difficile infection. PLoS Comput. Biol. 2021, 17, e1008782. [Google Scholar] [CrossRef] [PubMed]
- Phalak, P.; Henson, M.A. Metabolic Modeling of Clostridium difficile Associated Dysbiosis of the Gut Microbiota. Processes 2019, 7, 97. [Google Scholar] [CrossRef]
- Knippel, R.J.; Wexler, A.G.; Miller, J.M.; Beavers, W.N.; Weiss, A.; de Crécy-Lagard, V.; Edmonds, K.A.; Giedroc, D.P.; Skaar, E.P. Clostridioides difficile Senses and Hijacks Host Heme for Incorporation into an Oxidative Stress Defense System. Cell Host Microbe 2020, 28, 411–421.e6. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.R.; Pike, C.M.; Parsons, R.J.; Rivera, A.J.; Foley, M.H.; McLaren, M.R.; Montgomery, S.A.; Theriot, C.M. Clostridioides difficile Exploits Toxin-Mediated Inflammation to Alter the Host Nutritional Landscape and Exclude Competitors from the Gut Microbiota. Nat. Commun. 2021, 12, 462. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meza-Torres, J.; Auria, E.; Dupuy, B.; Tremblay, Y.D.N. Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections. Microorganisms 2021, 9, 1922. https://doi.org/10.3390/microorganisms9091922
Meza-Torres J, Auria E, Dupuy B, Tremblay YDN. Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections. Microorganisms. 2021; 9(9):1922. https://doi.org/10.3390/microorganisms9091922
Chicago/Turabian StyleMeza-Torres, Jazmin, Emile Auria, Bruno Dupuy, and Yannick D. N. Tremblay. 2021. "Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections" Microorganisms 9, no. 9: 1922. https://doi.org/10.3390/microorganisms9091922
APA StyleMeza-Torres, J., Auria, E., Dupuy, B., & Tremblay, Y. D. N. (2021). Wolf in Sheep’s Clothing: Clostridioides difficile Biofilm as a Reservoir for Recurrent Infections. Microorganisms, 9(9), 1922. https://doi.org/10.3390/microorganisms9091922