
Characterization of Martelella soudanensis sp. nov., Isolated from a Mine Sediment

Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrichment Culture and Isolation
2.2. Phenotypic Analysis
2.3. Chemotaxonomic Analysis
2.4. Phylogenetic Analysis
2.5. Whole-Genome Sequencing, Assembly, and Annotation
2.6. Comparative Genomic Analysis
3. Results and Discussion
3.1. Phenotypic Characterization
3.2. Chemotaxonomic Characterization
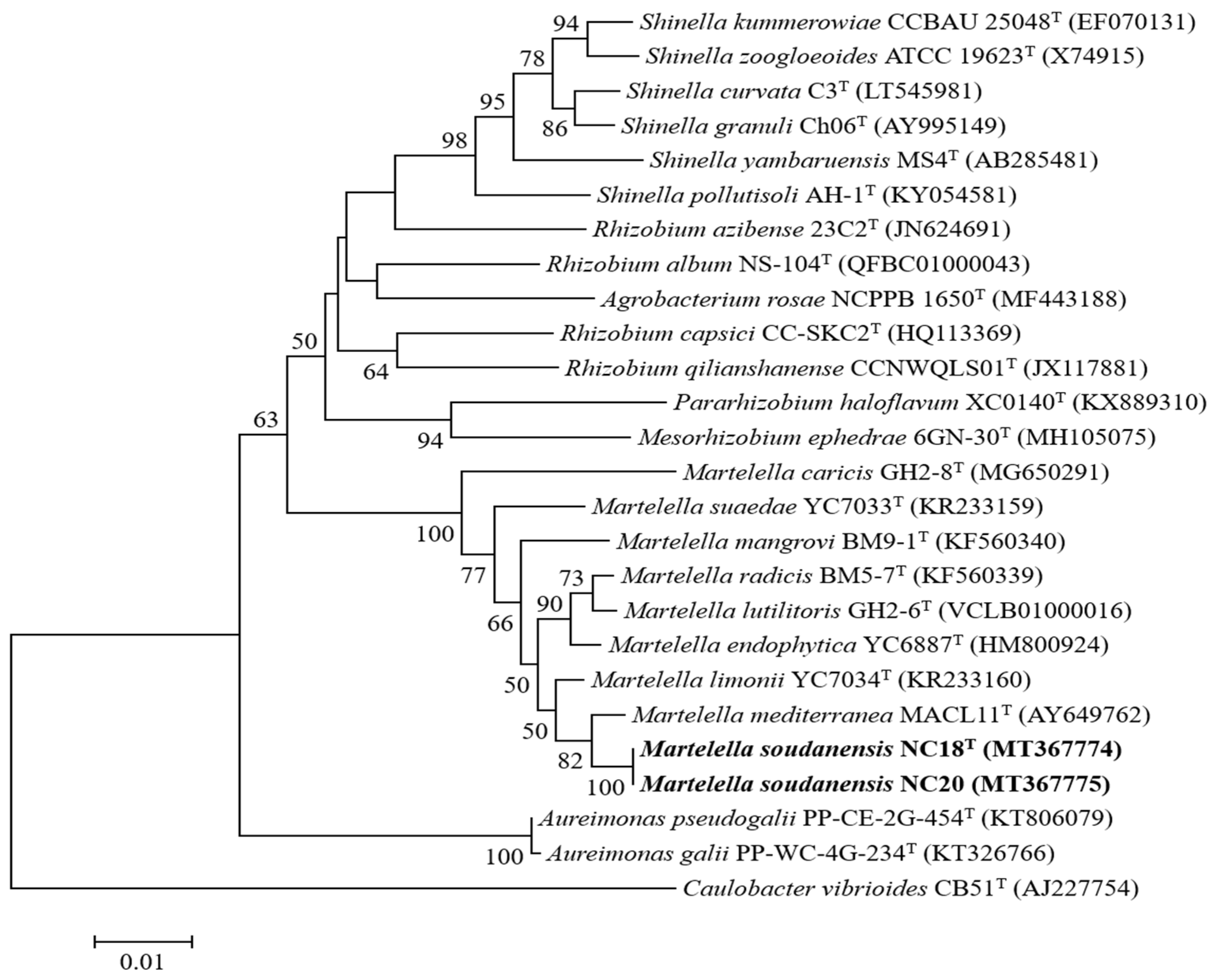
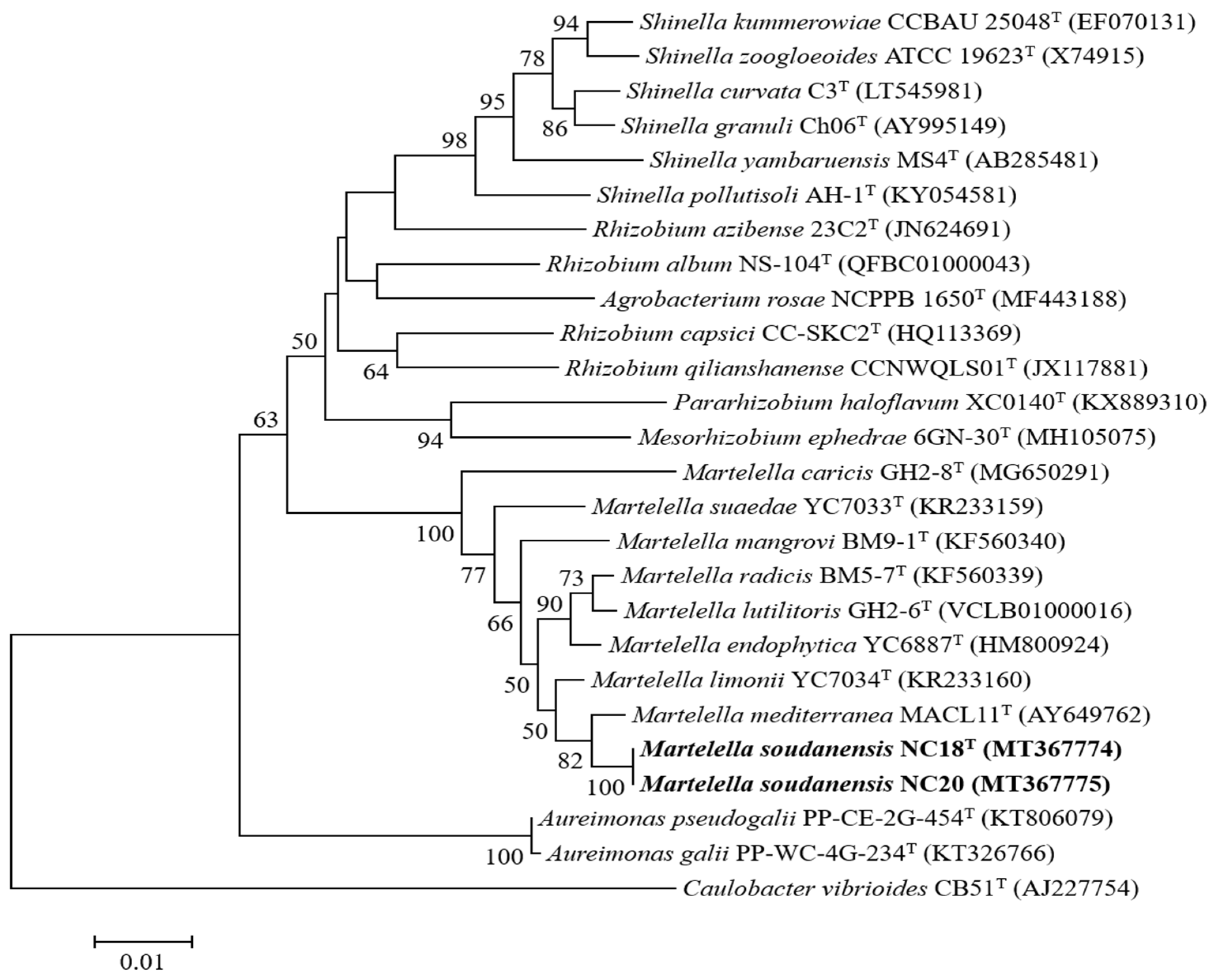
3.3. Phylogenetic Characterization
3.4. General Genomic Features
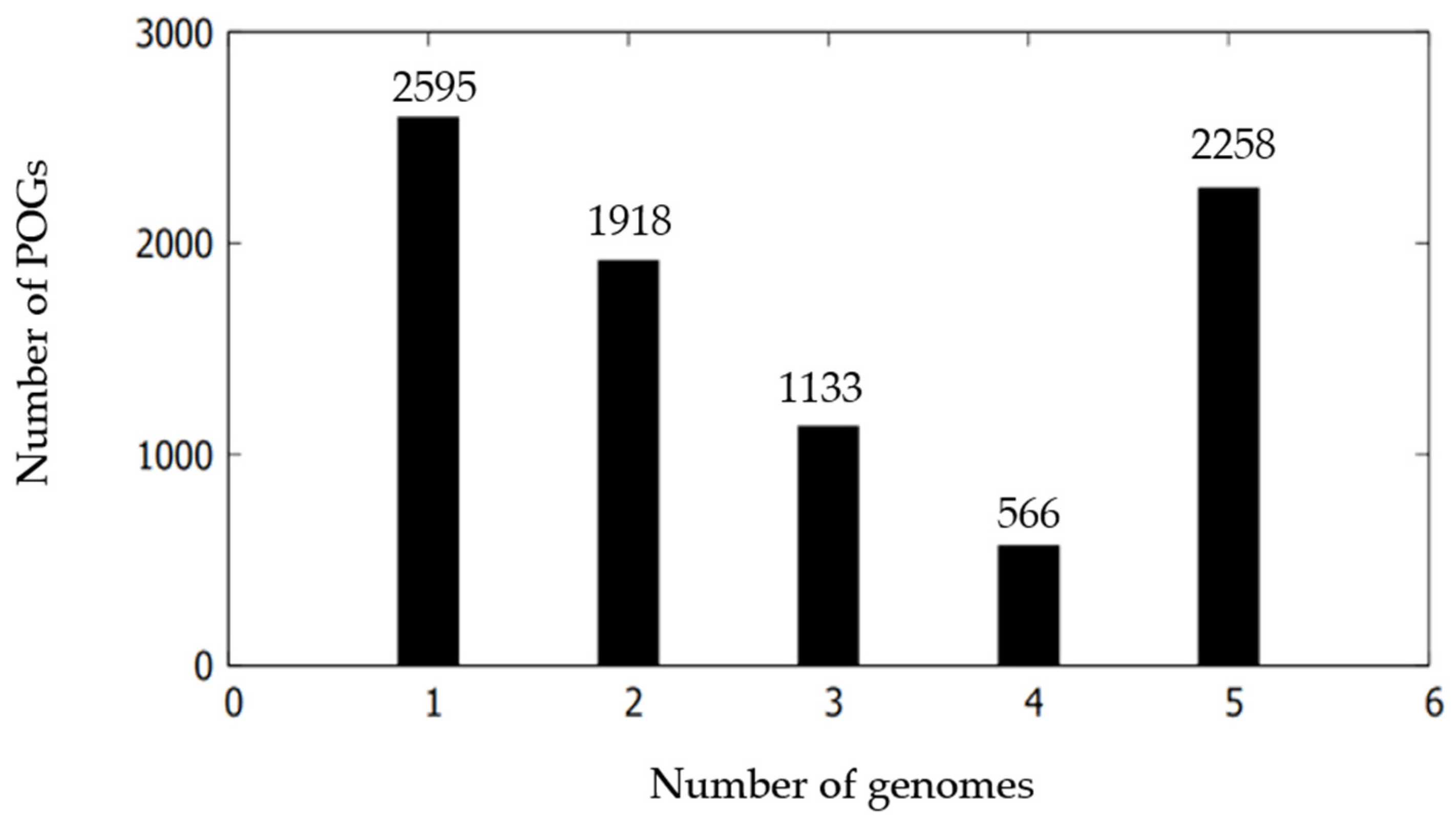
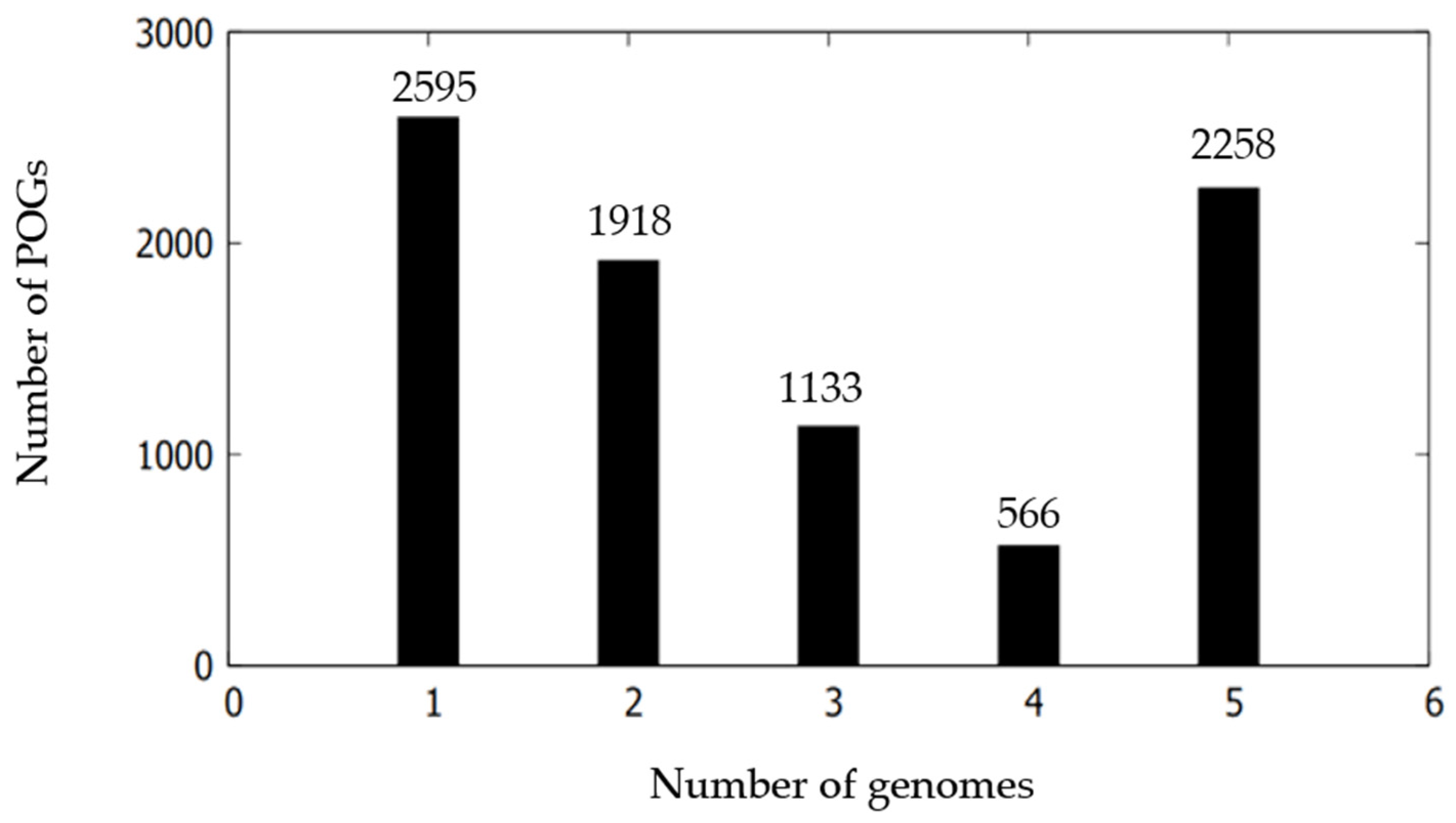
3.5. Comparative Genomic Characterization
4. Conclusions
4.1. Description of Martelella Soudanensis sp. nov.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kadnikov, V.V.; Mardanov, A.V.; Beletsky, A.V.; Karnachuk, O.V.; Ravin, N.V. Microbial Life in the Deep Subsurface Aquifer Illuminated by Metagenomics. Front. Microbiol. 2020, 11, 2146. [Google Scholar] [CrossRef]
- Breuker, A.; Köweker, G.; Blazejak, A.; Schippers, A. The deep biosphere in terrestrial sediments in the Chesapeake Bay area, Virginia, USA. Front. Microbiol. 2011, 2, 156. [Google Scholar] [CrossRef] [Green Version]
- Sheik, C.; Badalamenti, J.; Telling, J.; Hsu, D.; Alexander, S.C.; Bond, D.R.; Gralnick, J.A.; Sherwood Lollar, B.; Toner, B.M. Novel microbial groups drive productivity in an Archean Iron formation. Front. Microbiol. 2021, 12, 616. [Google Scholar] [CrossRef]
- Badalamenti, J.P.; Summers, Z.M.; Chan, C.H.; Gralnick, J.A.; Bond, D.R. Isolation and genomic characterization of ‘Desulfuromonas soudanensis WTL’, a metal-and electrode-respiring bacterium from anoxic deep subsurface brine. Front. Microbiol. 2016, 7, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonis, B.M.; Gralnick, J.A. Marinobacter subterrani, a genetically tractable neutrophilic Fe (II)-oxidizing strain isolated from the Soudan Iron Mine. Front. Microbiol. 2015, 6, 719. [Google Scholar] [CrossRef]
- Held, B.W.; Salomon, C.E.; Blanchette, R.A. Diverse subterranean fungi of an underground iron ore mine. PLoS ONE 2020, 15, e0234208. [Google Scholar] [CrossRef]
- Ventosa, A.; Márquez, M.C.; Sánchez-Porro, C.; Rafael, R. Taxonomy of halophilic archaea and bacteria. In Advances in Understanding the Biology of Halophilic Microorganisms; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–80. [Google Scholar]
- Fathepure, B.Z. Recent studies in microbial degradation of petroleum hydrocarbons in hypersaline environments. Front. Microbiol. 2014, 5, 173. [Google Scholar] [CrossRef] [Green Version]
- Corral, P.; Amoozegar, M.A.; Ventosa, A. Halophiles and their biomolecules: Recent advances and future applications in biomedicine. Mar. Drugs 2020, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Le Borgne, S.; Paniagua, D.; Vazquez-Duhalt, R. Biodegradation of organic pollutants by halophilic bacteria and archaea. J. Mol. Microbiol. Biotechnol. 2008, 15, 74–92. [Google Scholar] [CrossRef]
- Cai, Z.; Ge, H.; Yi, Z.; Zeng, R.; Zhang, G. Characterization of a novel psychrophilic and halophilic β-1, 3-xylanase from deep-sea bacterium, Flammeovirga pacifica strain WPAGA1. Int. J. Biol. Macromol. 2018, 118, 2176–2184. [Google Scholar] [CrossRef] [PubMed]
- Amoozegar, M.A.; Safarpour, A.; Noghabi, K.A.; Bakhtiary, T.; Ventosa, A. Halophiles and their vast potential in biofuel production. Front. Microbiol. 2019, 10, 1895. [Google Scholar] [CrossRef] [PubMed]
- Ruginescu, R.; Gomoiu, I.; Popescu, O.; Cojoc, R.; Neagu, S.; Lucaci, I.; Batrinescu-Moteau, C.; Enache, M. Bioprospecting for novel halophilic and halotolerant sources of hydrolytic enzymes in brackish, saline and hypersaline lakes of Romania. Microorganisms 2020, 8, 1903. [Google Scholar] [CrossRef] [PubMed]
- Giani, M.; Garbayo, I.; Vílchez, C.; Martínez-Espinosa, R.M. Haloarchaeal carotenoids: Healthy novel compounds from extreme environments. Mar. Drugs 2019, 17, 524. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Chen, J.; Wu, Q.; Chen, G. Halophiles, coming stars for industrial biotechnology. Biotechnol Adv. 2015, 33, 1433–1442. [Google Scholar] [CrossRef]
- Liu, C.; Baffoe, D.K.; Zhan, Y.; Zhang, M.; Li, Y.; Zhang, G. Halophile, an essential platform for bioproduction. J. Microbiol. Methods 2019, 166, 105704. [Google Scholar] [CrossRef]
- Oren, A. Industrial and environmental applications of halophilic microorganisms. Environ. Technol 2010, 31, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Oren, A. Diversity of halophilic microorganisms: Environments, phylogeny, physiology, and applications. J. Ind. Microbiol. Biotechnol. 2002, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rivas, R.; Sanchez-Marquez, S.; Mateos, P.F.; Martínez-Molina, E.; Velazquez, E. Martelella mediterranea gen. nov., sp. nov., a novel α-proteobacterium isolated from a subterranean saline lake. Int. J. Syst. Evol. Microbiol. 2005, 55, 955–959. [Google Scholar] [CrossRef]
- Bibi, F.; Chung, E.J.; Khan, A.; Jeon, C.O.; Chung, Y.R. Martelella endophytica sp. nov., an antifungal bacterium associated with a halophyte. Int. J. Syst. Evol. Microbiol. 2013, 63, 2914–2919. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Margesin, R. Martelella radicis sp. nov. and Martelella mangrovi sp. nov., isolated from mangrove sediment. Int. J. Syst. Evol. Microbiol. 2014, 64, 3104–3108. [Google Scholar] [CrossRef]
- Chung, E.J.; Hwang, J.M.; Kim, K.H.; Jeon, C.O.; Chung, Y.R. Martelella suaedae sp. nov. and Martelella limonii sp. nov., isolated from the root of halophytes. Int. J. Syst. Evol. Microbiol. 2016, 66, 3917–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D. Martelella caricis sp. nov., isolated from a rhizosphere mudflat. Int. J. Syst. Evol. Microbiol. 2019, 69, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S.D. Martelella lutilitoris sp. nov., isolated from a tidal mudflat. J. Microbiol. 2019, 57, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Ma, L.; Shi, J.; Lin, K.; Luo, Q.; Liu, Y. Metabolic pathway for degradation of anthracene by halophilic Martelella sp. AD-3. Int. Biodeterior. Biodegrad. 2014, 89, 67–73. [Google Scholar] [CrossRef]
- Sasser, M. Identification of Bacteria by Gas Chromatography of Cellular Fatty Acids; MIDI, Inc.: Newark, Delaware, 1990. [Google Scholar]
- Shin, Y.K.; Lee, J.; Lee, K.C.; Chun, C.O.; Kim, H.; Joo, W.; Lee, J.; Park, Y. Isoprenoid quinone profiles in microbial taxonomy. J. Life Sci. 1995, 5, 211–217. [Google Scholar]
- Weisburg, W.G.; Barns, S.M.; Pelletier, D.A.; Lane, D.J. 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 1991, 173, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Ha, S.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. Gerf. Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Version 6.0. Molecular evolutionary genetics analysis. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Kerepesi, C.; Bánky, D.; Grolmusz, V. AmphoraNet: The webserver implementation of the AMPHORA2 metagenomic workflow suite. Gene 2014, 533, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Lee, I.; Chalita, M.; Ha, S.; Na, S.; Yoon, S.; Chun, J. ContEst16S: An algorithm that identifies contaminated prokaryotic genomes using 16S RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2017, 67, 2053–2057. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Burge, S.W.; Bateman, A.; Daub, J.; Eberhardt, R.Y.; Eddy, S.R.; Floden, E.W.; Gardner, P.P.; Jones, T.A.; Tate, J. Rfam 12.0: Updates to the RNA families database. Nucleic Acids Res. 2015, 43, D130–D137. [Google Scholar] [CrossRef]
- Edgar, R.C. PILER-CR: Fast and accurate identification of CRISPR repeats. BMC Bioinform. 2007, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldon, T.; Rattei, T.; Creevey, C.; Kuhn, M. eggNOG v4. 0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014, 42, D231–D239. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, L.M.; Konstantinidis, K.T. Bypassing cultivation to identify bacterial species. Microbe 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.; De Meyer, S. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Characteristic | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Cell size (μm) | 0.8–0.9 × 1.2–1.3 | 0.6–0.8 × 1.1–1.3 | 1.0–1.1 × 1.3–1.5 | 0.8–1.1 × 1.2–2.5 | 0.5–0.8 × 1.2–2.3 | 1.0 × 1.2 | 0.8–0.9 × 1.9–2.8 | 0.4–0.7 × 1.0–1.5 | 1.0–1.2 × 1.2–1.5 | 0.8–0.9 × 1.3–1.4 |
| Conditions for growth: | ||||||||||
| Temperature (°C) | 10–40 (30) | 4–35 (30) | 4–37 (28) | 4–40 (25–30) | 4–40 (28–30) | 10–40 | 20–45 (30) | 10–40 (25–30) | 10–40 | 10–30 (30) |
| pH | 5–9 (7) | 5–9 (8) | 5–8 (7) | 5–9 (6–8.5) | 5–9 (7–8.5) | 5–9.5 | 5–10 (6–8) | 5–10 (7–8) | 5–8 | 4–10 (7–8) |
| NaCl (%) | 0–13 (1) | 0–13 (1) | 0–5 (2) | 1–6 (2–5) | 0–9 (4–5) | 2–12 | 0.5–9 (0.5–3) | 1–9 (2–6) | 2–10 | 0–11 (0–2) |
| Nitrate reduction | + | + | + | NR | + | - | - | NR | + | - |
| Aesculin hydrolysis | - | - | + | + | + | + | w | + | + | + |
| acetoin production | - | - | + | + | + | NR | NR | + | NR | NR |
| Fermentation-oxidation: | ||||||||||
| L-rhamnose | + | + | - | - | - | NR | NR | - | NR | NR |
| D-glucose | - | - | NR | - | - | + | + | - | + | NR |
| D-mannitol | - | - | NR | - | - | NR | + | - | NR | NR |
| L-arabinose | + | + | NR | - | + | NR | + | + | NR | NR |
| Assimilation: | ||||||||||
| potassium gluconate | - | - | + | - | - | - | - | + | - | - |
| malate | - | - | + | - | - | + | - | + | - | - |
| D-maltose | - | - | + | - | + | - | - | + | + | - |
| D-mannose | - | - | - | - | + | NR | - | + | NR | - |
| phenylacetate | - | - | - | NR | - | + | - | NR | + | - |
| D-glucose | - | - | + | - | + | + | - | + | + | - |
| D-mannitol | - | - | - | - | + | + | - | + | + | - |
| L-arabinose | - | - | - | - | + | NR | - | + | w | - |
| Enzyme activity: | ||||||||||
| Urease | + | + | + | - | - | - | - | - | - | + |
| esterase lipase (C8) | - | - | + | + | - | + | + | + | + | + |
| lipase (C14) | - | - | + | - | - | - | - | - | - | - |
| trypsin | - | - | + | + | - | - | - | + | - | - |
| naphthol-AS-BI-phosphohydrolase | + | + | - | w | - | w | + | - | + | + |
| α-galactosidase | - | - | + | - | - | w | - | w | - | - |
| β-glucuronidase | - | - | + | - | - | - | - | + | - | - |
| β-glucosidase | + | - | + | - | + | + | + | + | + | + |
| N-acetyl-β-glucosaminidase | + | - | + | - | + | - | - | - | - | + |
| α-fucosidase | - | - | - | - | + | - | - | - | - | - |
| G + C content (%) | 61.8 | 61.8 | 57.4 | 62.2 | 62.1 | 59.7 | 61.9 | 52.8 | 61 | 53.4 |
| Fatty Acid | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Saturated: | ||||||||||
| C16:0 | 13.0 | 13.4 | 12.0 | 8.4 | 4.8 | 5.9 | 10.0 | 7.8 | 2.5 | 17.2 |
| C17:0 | 0.8 | 0.7 | - | 1.1 | - | 1.2 | <1 | 1.2 | - | - |
| C18:0 | 6.2 | 6.3 | 4.3 | 6.9 | 8.8 | 8.9 | 10.4 | 6.6 | 7.6 | 2.9 |
| C19:0 cyclo ω8c | 25.8 | 28.2 | 41.4 | 25.3 | 28.0 | 23.7 | 9.8 | 20.8 | 24.9 | 12.5 |
| Branched: | ||||||||||
| C18:1 ω7c | - | - | 21.7 | - | 17.9 | 35.7 | - | - | 41.7 | - |
| C18:1 ω5c | 0.4 | 0.4 | - | - | - | - | - | - | - | - |
| C18:1 ω7c 11-methyl | 6.4 | 6.6 | 8.8 | 17.8 | 7.9 | 5.4 | 8.4 | 16.2 | 6.8 | 8.9 |
| C19:0 iso | 0.3 | - | - | - | - | - | - | - | - | - |
| Unsaturated: | ||||||||||
| C20:1 ω7c | 0.3 | 0.3 | - | - | - | - | - | - | - | - |
| C20:2 ω6,9c | 0.6 | 0.7 | 0.7 | - | - | - | - | - | - | 1.4 |
| Hydroxy: | ||||||||||
| C16:0 3-OH | 0.7 | 0.7 | 0.5 | 0.3 | - | - | <1 | 0.3 | - | 0.9 |
| C18:1 2-OH | 0.2 | - | - | 0.5 | - | 1.5 | <1 | 0.5 | 1.9 | 0.9 |
| C18:0 3-OH | 1.0 | 1.0 | 0.7 | 0.5 | <1 | 0.7 | <1 | 0.4 | 0.7 | 0.5 |
| Summed feature* | ||||||||||
| 2 | 5.9 | 5.6 | 7.7 | 14.6 | 9.6 | 14.8 | 7.5 | 14.5 | 12.4 | 8.8 |
| 3 | 0.5 | 0.5 | 0.6 | 0.5 | - | - | 1.3 | 0.5 | - | 1.9 |
| 7 | 0.5 | 0.6 | - | - | - | - | - | 0.5 | - | 2.5 |
| 8 | 37.5 | 35.0 | - | 23.4 | - | - | 35.9 | 30.2 | - | 39.7 |
| Feature | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| Genome size (bp) | 6,109,459 | 6,109,677 | 5,693,067 | 4,817,335 | 4,447,501 |
| N50 (bp) | 5,586,623 | 5,586,823 | 4,671,477 | 4,817,335 | 402,005 |
| Contig number | 3 | 3 | 4 | 1 | 18 |
| Assembly level | complete | complete | complete | complete | contig |
| Total genes | 5849 | 5830 | 5303 | 4539 | 4093 |
| Protein coding genes | 5502 | 5585 | 5115 | 4409 | 3981 |
| rRNA | 6 | 6 | 6 | 9 | 3 |
| tRNA | 48 | 48 | 48 | 53 | 47 |
| GenBank assembly accession number | GCA_013459415.1 | GCA_013459645.1 | GCA_002043005.1 | GCA_000960975.1 | GCA_005924265.1 |
| Strain | ANI Values (%) | dDDH Values (%) | ||
|---|---|---|---|---|
| NC18T | NC20 | NC18T | NC20 | |
| Martelella soudanensis NC18T | - | 99.9 | - | 100 |
| Martelella soudanensis NC20 | 99.9 | - | 100 | - |
| Martelella mediterranea DSM 17316T | 88.1 | 88.0 | 34.9 | 34.9 |
| Martelella endophytica YC6887T | 80.2 | 80.4 | 23.9 | 23.9 |
| Martelella lutilitoris GH2-6T | 80.4 | 80.4 | 23.7 | 23.7 |
| Strains | M. soudanensis NC18T | M. soudanensis NC20 | M. mediterranea DSM 17316T | M. endophytica YC6887T | M. lutilitoris GH2-6T |
|---|---|---|---|---|---|
| M. soudanensis NC18T | 100 | 87.7 | 76.3 | 77.8 | |
| M. soudanensis NC20 | 100 | 87.9 | 76.5 | 78.0 | |
| M. mediterranea DSM 17316T | 87.7 | 87.9 | 77.3 | 78.2 | |
| M. endophytica YC6887T | 76.3 | 76.5 | 77.3 | 78.5 | |
| M. lutilitoris GH2-6T | 77.8 | 78.0 | 78.2 | 78.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-Y.; Lee, D.-H.; Kim, D.-H. Characterization of Martelella soudanensis sp. nov., Isolated from a Mine Sediment. Microorganisms 2021, 9, 1736. https://doi.org/10.3390/microorganisms9081736
Lee J-Y, Lee D-H, Kim D-H. Characterization of Martelella soudanensis sp. nov., Isolated from a Mine Sediment. Microorganisms. 2021; 9(8):1736. https://doi.org/10.3390/microorganisms9081736
Chicago/Turabian StyleLee, Jung-Yun, Dong-Hun Lee, and Dong-Hun Kim. 2021. "Characterization of Martelella soudanensis sp. nov., Isolated from a Mine Sediment" Microorganisms 9, no. 8: 1736. https://doi.org/10.3390/microorganisms9081736
APA StyleLee, J.-Y., Lee, D.-H., & Kim, D.-H. (2021). Characterization of Martelella soudanensis sp. nov., Isolated from a Mine Sediment. Microorganisms, 9(8), 1736. https://doi.org/10.3390/microorganisms9081736