
Repurposing of the Fasciolicide Triclabendazole to Treat Infections Caused by Staphylococcus spp. and Vancomycin-Resistant Enterococci

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antimicrobial Agents
2.2. Bacterial Strains
2.3. Minimum Inhibitory Concentration (MIC) Determination
2.4. Synergy Testing by Checkerboard Microdilution and Dose Reduction Analysis
2.5. Minimum Bactericidal Concentration (MBC) Determination
2.6. Time-Dependent Killing Assays
2.7. Multi-Sub-Culture Resistance Selection
2.8. Intracellular MIC Testing
2.9. Haemolysis Assay
2.10. In Vitro Cytotoxicity Assays
2.11. Ethics Statements
2.12. Oral Safety Assessment of TCBZ following Parenteral Administration
2.13. Histopathological Examination
2.14. Oral Efficacy Testing of TCBZ following Systemic Challenge of Mice with Bioluminescent Gram-Positive Bacteria
3. Results
3.1. TCBZ Derivatives Demonstrate Antibacterial Activity
3.2. TCBZ Shows Antimicrobial Activity against an Expanded Range of Staphylococcus spp. and Vancomycin-Resistant Enterococci
3.3. TCBZ Shows Limited Antimicrobial Activity against Streptococcus spp. Causing Bovine Mastitis
3.4. TCBZ in Combination with PMB Demonstrates Synergistic Activity against a Range of Gram-Negative ESKAPE Pathogens
3.5. TCBZ Kinetic Assays Confirm Bactericidal Activity
3.6. No TCBZ-Resistant Mutants Developed after 21 Daily Sequential In Vitro Sub-Cultures
3.7. Intracellular MIC Testing of TCBZ Shows Bacteriostatic Activity against S. aureus but Bactericidal Activity against MRSP
3.8. TCBZ Is Non-Hemolytic and Non-Cytotoxic
3.9. TCBZ Shows Oral Safety in Mice
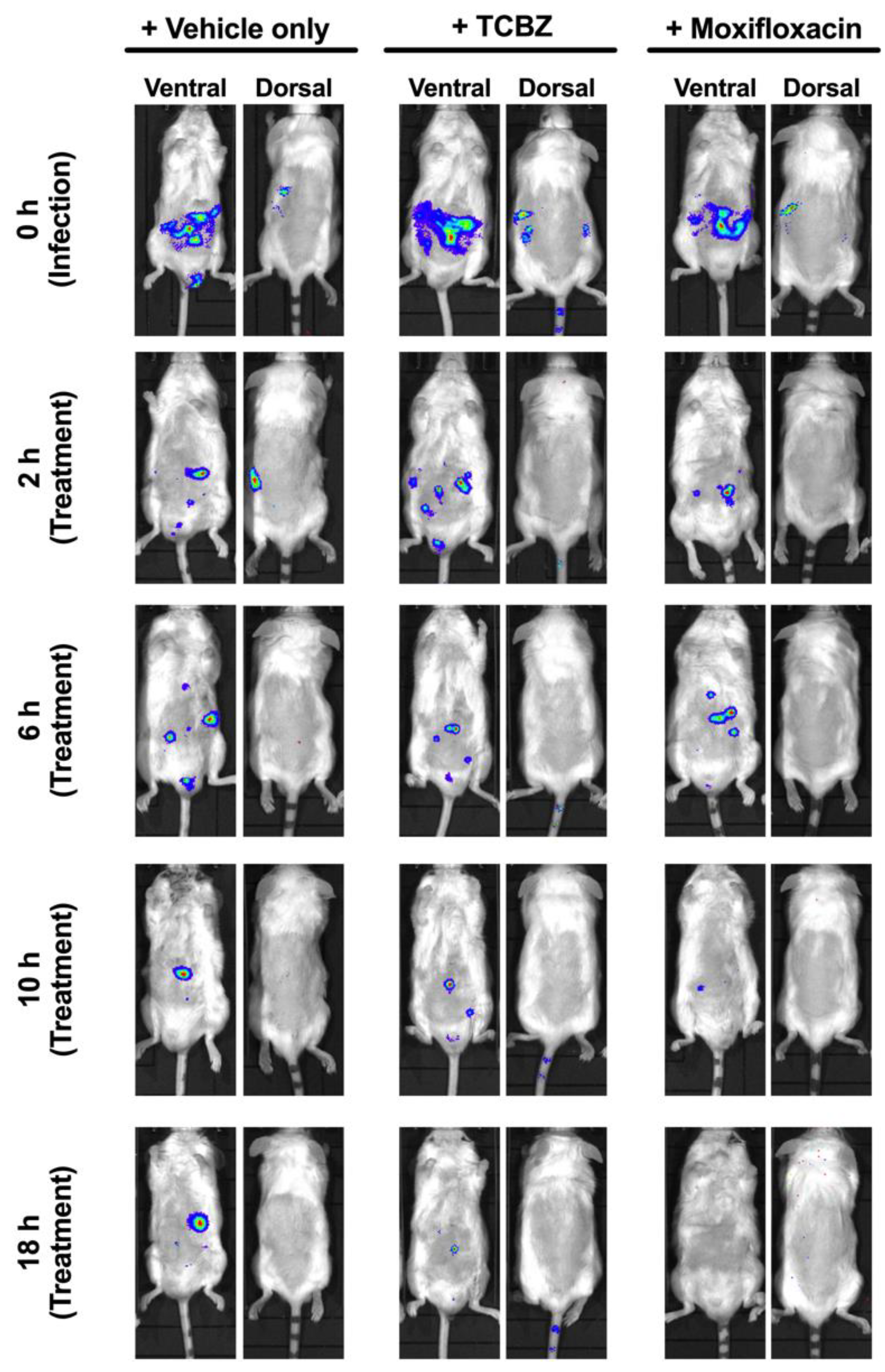
3.10. Treatment of Mice with TCBZ Reduces S. aureus Population with Repeated Treatments within 24 h Post-Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Hong, I.S.; Ipema, H.J.; Gabay, M.P.; Lodolce, A.E. Medication Repurposing: New Uses for Old Drugs. J. Pharm Tech. 2011, 27, 132–140. [Google Scholar] [CrossRef]
- O’Neill, J.; Davies, S.; Rex, J.; White, L.; Murray, R. Review on antimicrobial resistance, tackling drug-resistant infections globally: Final report and recommendations. Lond. Wellcome Trust. UK Gov. 2016, 1, 84. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41. [Google Scholar] [CrossRef]
- Australian Commission on Safety and Quality in Health Care. Antimicrobial Stewardship in Australian Health Care 2018; ACSQHC: Sydney, Australia, 2018. [Google Scholar]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Brown, D. Antibiotic resistance breakers: Can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015, 14, 821–832. [Google Scholar] [CrossRef]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharm. 2018, 175, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprea, T.I.; Mestres, J. Drug repurposing: Far beyond new targets for old drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Strittmatter, S.M. Overcoming drug development bottlenecks with repurposing: Old drugs learn new tricks. Nat. Med. 2014, 20, 590–591. [Google Scholar] [CrossRef] [Green Version]
- Durand, G.A.; Raoult, D.; Dubourg, G. Antibiotic discovery: History, methods and perspectives. Int. J. Antimicrob. Agents 2019, 53, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano-Tinoco, E.A.; García-Contreras, S.J.; García-Contreras, R. Drug repurposing as an alternative for the treatment of recalcitrant bacterial infections. Front. Microbiol. 2015, 6, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharm. 2018, 175, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Chowdhury, E.H.; Hossain, M.I.; Huque, A.K.M.F.; Mondal, M.M.H. The Efficacy of the Triclabendazole (Fasinex AE) against Fasciola gigantica infection in cattle of Bangladesh. Prog. Agric. 2001, 12, 151–155. [Google Scholar]
- El-Tantawy, W.H.; Salem, H.F.; Mohammed Safwat, N.A. Effect of Fascioliasis on the pharmacokinetic parameters of triclabendazole in human subjects. Pharm. World Sci. 2007, 29, 190–198. [Google Scholar] [CrossRef]
- Lecaillon, J.B.; Godbillon, J.; Campestrini, J.; Naquira, C.; Miranda, L.; Pacheco, R.; Mull, R.; Poltera, A.A. Effect of food on the bioavailability of triclabendazole in patients with fascioliasis. Br. J. Clin. Pharm. 1998, 45, 601–604. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food & Drug Adminstration. New Drug Therapy Approvals 2019. WHO/MVP/EMP/IAU/2019.07. 2020. Available online: https://www.fda.gov/media/134493/download (accessed on 29 July 2021).
- World Health Organization. World Health Organization Model List of Essential Medicines 21st List. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/325771/WHO-MVP-EMP-IAU-2019.06-eng.pdf (accessed on 29 July 2021).
- World Health Organization. World Health Organization Model List of Essential Medicines for Children: 7th List 2019. 2019. Available online: https://apps.who.int/iris/bitstream/handle/10665/325772/WHO-MVP-EMP-IAU-2019.07-eng.pdf (accessed on 29 July 2021).
- Younis, W.; Thangamani, S.; Seleem, M.N. Repurposing non-antimicrobial drugs and clinical molecules to treat bacterial infections. Curr. Pharm. Des. 2015, 21, 4106–4111. [Google Scholar] [CrossRef] [Green Version]
- AbdelKhalek, A.; Mohammad, H.; Mayhoub, A.S.; Seleem, M.N. Screening for potent and selective anticlostridial leads among FDA-approved drugs. J. Antibiot. 2020, 73, 392–409. [Google Scholar] [CrossRef]
- Schneider, E.K.; Reyes-Ortega, F.; Velkov, T.; Li, J. Antibiotic-non-antibiotic combinations for combating extremely drug-resistant Gram-negative ‘superbugs’. Essays Biochem. 2017, 61, 115–125. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Löpez, C.A.; Gnanakaran, S. Permeability barrier of Gram-negative cell envelopes and approaches to bypass it. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Pi, H.; Nguyen, H.T.; Venter, H.; Boileau, A.R.; Woolford, L.; Garg, S.; Page, S.W.; Russell, C.C.; Baker, J.R.; McCluskey, A.; et al. In vitro activity of robenidine analog ncl195 in combination with outer membrane permeabilizers against Gram-negative bacterial pathogens and impact on systemic Gram-positive bacterial infection in mice. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Lynch, B.M.; Chen, C.M.; Wigfield, Y.-Y. Nitrations of acetanilides by reagents of N02X type. Can. J. Chem. 1968, 46, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- van Allan, J.A.; Deacon, B.D. 2-Mercaptobenzimidazole; John Wiley & Sons: New York, NY, USA, 1963; Volume lV. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/2-Mercaptobenzimidazole (accessed on 29 July 2021).
- Averkin, E.A.; Beard, C.C.; Dvorak, C.A.; Edwards, J.A.; Fried, J.H.; Kilian, J.G.; Schiltz, R.A.; Kistner, T.P.; Drudge, J.H.; Ly-ons, E.T.; et al. Methyl 5(6)-phenylsulfinyl-2-benzimidazole-carbamate, a new, potent anthelmintic. J. Med. Chem. 1975, 18, 1164–1166. [Google Scholar] [CrossRef]
- Vandenberk, J.; Kennis, L.E.J.; Van de Aa, M.J.M.C.; Van Heertum, A.H.M.T. Antiemetic 1-(benzimidazolyalkyl) Piperidine Derivatives. 1978, Volume 4. Available online: https://patents.google.com/patent/US4126687A/en (accessed on 29 July 2021).
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; CLSI Standard M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Venter, H. Reversing resistance to counter antimicrobial resistance in the World Health Organisation’s critical priority of most dangerous pathogens. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamoud, R.; Reichling, J.; Wink, M. Synergistic antibacterial activity of the combination of the alkaloid sanguinarine with EDTA and the antibiotic streptomycin against multidrug resistant bacteria. J. Pharm. Pharm. 2015, 67, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Khazandi, M.; Pi, H.; Chan, W.Y.; Ogunniyi, A.D.; Sim, J.X.F.; Venter, H.; Garg, S.; Page, S.W.; Hill, P.B.; McCluskey, A. In vitro antimicrobial activity of robenidine, ethylenediaminetetraacetic acid and polymyxin B nonapeptide against important human and veterinary pathogens. Front. Microbiol. 2019, 10, 837. [Google Scholar] [CrossRef] [Green Version]
- Eid, S.Y.; El-Readi, M.Z.; Wink, M. Synergism of three-drug combinations of sanguinarine and other plant secondary metabolites with digitonin and doxorubicin in multi-drug resistant cancer cells. Phytomed 2012, 19, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Ogunniyi, A.D.; Khazandi, M.; Stevens, A.J.; Sims, S.K.; Page, S.W.; Garg, S.; Venter, H.; Powell, A.; White, K.; Petrovski, K.R.; et al. Evaluation of robenidine analog NCL195 as a novel broad-spectrum antibacterial agent. PLoS ONE 2017, 12, e0183457. [Google Scholar] [CrossRef] [Green Version]
- Seral, C.; Van Bambeke, F.; Tulkens, P.M. Quantitative analysis of gentamicin, azithromycin, telithromycin, ciprofloxacin, moxifloxacin, and oritavancin (LY333328) activities against intracellular Staphylococcus aureus in mouse J774 macrophages. Antimicrob Agents Chemother. 2003, 47, 2283–2292. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.-P.; Lee, W.; Tan, T.-Y.; Sasikala, S.; Teo, J.; Hsu, L.-Y.; Tan, T.-T.; Syahidah, N.; Kwa, A.L. Effective antibiotics in combination against extreme drug-resistant Pseudomonas aeruginosa with decreased susceptibility to polymyxin B. PLoS ONE 2011, 6, e28177. [Google Scholar] [CrossRef]
- Ogunniyi, A.D.; Kopecki, Z.; Hickey, E.E.; Khazandi, M.; Peel, E.; Belov, K.; Boileau, A.; Garg, S.; Venter, H.; Chan, W.Y.; et al. Bioluminescent murine models of bacterial sepsis and scald wound infections for antimicrobial efficacy testing. PLoS ONE 2018, 13, e0200195. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis. 2021. Available online: https://apps.who.int/iris/handle/10665/340694 (accessed on 29 July 2021).
- The PEW Charitable Trusts. Tracking the Global Pipeline of Antibiotics in Development, March 2021. Available online: https://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2021/03/tracking-the-global-pipeline-of-antibiotics-in-development (accessed on 29 July 2021).
- Bassetti, M.; Peghin, M.; Vena, A.; Giacobbe, D.R. Treatment of infections due to MDR Gram-negative bacteria. Front. Med. 2019, 6. [Google Scholar] [CrossRef]
- Theuretzbacher, U. Global antimicrobial resistance in Gram-negative pathogens and clinical need. Curr. Opin. Microbiol. 2017, 39, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kelley, J.M.; Elliott, T.P.; Beddoe, T.; Anderson, G.; Skuce, P.; Spithill, T.W. Current threat of triclabendazole resistance in Fasciola hepatica. Trends Parasitol. 2016, 32, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Advisory Group on Integrated Surveillance of Antimicrobial Resistance (AGISAR): Critically Important Antimicrobials for Human Medicine 6th Revision 2018. 2019. Available online: https://www.who.int/publications/i/item/9789241515528 (accessed on 29 July 2021).
- Roberts, K.D.; Azad, M.A.; Wang, J.; Horne, A.S.; Thompson, P.E.; Nation, R.L.; Velkov, T.; Li, J. Antimicrobial activity and toxicity of the major lipopeptide components of polymyxin B and colistin: Last-line antibiotics against multidrug-resistant Gram-negative bacteria. ACS Infect. Dis. 2015, 1, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, P.; Schmitt, E.K.; Chen, C.W.; Samantray, S.; Venishetty, V.K.; Hughes, D. Triclabendazole in the treatment of human fascioliasis: A review. Trans. R. Soc. Trop. Med. Hyg. 2019, 113, 797–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetzstein, H.-G. Comparative mutant prevention concentrations of pradofloxacin and other veterinary fluoroquinolones indicate differing potentials in preventing selection of resistance. Antimicrob Agents Chemother. 2005, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.U.; Velkov, T.; Lin, Y.-W.; Yun, B.; Nowell, C.J.; Zhou, F.; Zhou, Q.T.; Chan, K.; Azad, M.A.K.; Li, J. Potential Toxicity of Polymyxins in Human Lung Epithelial Cells. Antimicrob Agents Chemother. 2017, 61, e02690-16. [Google Scholar] [CrossRef] [Green Version]
- Algorri, M.; Wong-Beringer, A. Differential effects of antibiotics on neutrophils exposed to lipoteichoic acid derived from Staphylococcus aureus. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 50. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Malachowa, N.; DeLeo, F.R. Neutrophils and bacterial immune evasion. J. Innate Immun. 2018, 10, 432–441. [Google Scholar] [CrossRef]
- Bongers, S.; Hellebrekers, P.; Leenen, L.P.H.; Koenderman, L.; Hietbrink, F. Intracellular penetration and effects of antibiotics on Staphylococcus aureus inside human neutrophils: A comprehensive review. Antibiotics 2019, 8, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Identity | MIC (µg/mL) | |||
|---|---|---|---|---|---|
| VRE 60FR | VRE 252 | ATCC 49775 (MSSA) | USA300 (MRSA) | ||
| TCBZ * | Triclabendazole, 2-methyl thio | 4 | 8 | 2 | 2 |
| TCBZ-SO * | Triclabendazole, 2-methylsulphoxide | 16 | 16 | 8 | 8 |
| TCBZ-SO2 | Triclabendazole, 2-methylsulphone | >256 | >256 | 8 | >256 |
| TCBZ-SH * | Triclabendazole, 2 thio | 16 | 8 | 2 | 4 |
| TCBZ-OH | Triclabendazole, 2-hydroxy | >256 | >256 | >256 | >256 |
| Ampicillin | Ampicillin | 0.125 | 0.5 | <0.125 | 64 |
| Bacterial Strain/Isolate | MIC (µg/mL) for: | ||
|---|---|---|---|
| TCBZ | TCBZ-SO | TCBZ-SH | |
| VRE35C | 4 | 32 | 16 |
| VRE60FR | 8 | 32 | 16 |
| VRE252 | 4 | 16 | 16 |
| MRSA USA 300 | 2 | 8 | 2 |
| MSSA 49775 | 2 | 8 | 2 |
| MRSA 610 | 2 | 16 | 2 |
| S. pneumoniaeA66.1 | 16 | >64 | >64 |
| S. pneumoniaeD39 | 16 | >64 | >64 |
| Compound | Concentration (µg/mL) | |||
|---|---|---|---|---|
| MIC range | MIC50 | MIC90 | MBC | |
| TCBZ | 2–4 | 2 | 4 | 2–16 |
| Daptomycin | 0.25–1 | 0.5 | 0.5 | ND |
| Isolates | MIC (μg/mL) | Combination Effect (FICI) a | DRI b PMB: TCBZ | ||
|---|---|---|---|---|---|
| Single Drug | Combination | ||||
| PMB | TCBZ | PMB: TCBZ | |||
| E. coli ATCC 10763 | 0.5 | >256 | 0.125:0.25 | Synergism (0.25) | 4:1024 |
| E. coli ATCC 25922 | 0.5 | >256 | 0.125:0.125 | Synergism (0.25) | 4:2048 |
| P. aeruginosa PAO1 | 0.5 | >256 | 0.125:2 | Synergism (0.25) | 4:128 |
| K. pneumoniae ATCC 33495 | 0.5 | >256 | 0.125:1 | Synergism (0.25) | 4:256 |
| K. pneumoniae ATCC 4352 | 0.5 | >256 | 0.125:1 | Synergism (0.25) | 4:256 |
| A. baumannii ATCC 19606 | 1 | >256 | 0.125:0.5 | Synergism (0.125) | 8:512 |
| A. baumannii NCIMB 12457 | 1 | >256 | 0.125:1 | Synergism (0.125) | 8:256 |
| N. meningitidis 423 | >256 | 32 | 4:16 | Additivity (0.516) | 64:2 |
| N. meningitidis 424 | >256 | 32 | 4:16 | Additivity (0.516) | 64:2 |
| N. gonorrhoeae ATCC 16599 | >256 | >256 | 4:32 | Additivity (0.516) | 64:8 |
| N. gonorrhoeae ATCC 49226 | >256 | >256 | 4:32 | Additivity (0.516) | 64:8 |
| Isolates | Values | Antimicrobial Concentration (μg/mL) | Combination Effect (FICI) a | DRI b | ||||
|---|---|---|---|---|---|---|---|---|
| Single Drug | Combination | |||||||
| PMB | TCBZ | PMB | TCBZ | PMB | TCBZ | |||
| E. coli (n = 20) | MIC range | 0.125–1 | >256 | 0.06–0.125 | 0.25–2 | 2–8 | 128–2048 | |
| MIC50 | 0.5 | >256 | 0.125 | 0.5 | 4 | 512 | ||
| MIC90 | 0.5 | >256 | 0.125 | 1 | Synergism (0.25) | 4 | 256 | |
| K. pneumoniae (n = 20) | MIC range | 0.125–1 | >256 | 0.06–0.5 | 0.5–16 | 2–8 | 16–512 | |
| MIC50 | 0.5 | >256 | 0.25 | 4 | 2 | 64 | ||
| MIC90 | 1 | >256 | 0.5 | 8 | Additivity (0.53) | 2 | 32 | |
| A. baumannii (n = 18) | MIC range | 0.5–1 | >256 | 0.125–0.125 | 0.5–2 | 4–8 | 128–512 | |
| MIC50 | 1 | >256 | 0.125 | 2 | 8 | 128 | ||
| MIC90 | 1 | >256 | 0.125 | 2 | Synergism (0.13) | 8 | 128 | |
| P. aeruginosa (n = 20) | MIC range | 0.25–1 | >256 | 0.06–0.25 | 0.125–4 | 2–4 | 64–2048 | |
| MIC50 | 0.5 | >256 | 0.125 | 2 | 4 | 128 | ||
| MIC90 | 0.5 | >256 | 0.25 | 4 | Additivity (0.516) | 2 | 64 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pi, H.; Ogunniyi, A.D.; Savaliya, B.; Nguyen, H.T.; Page, S.W.; Lacey, E.; Venter, H.; Trott, D.J. Repurposing of the Fasciolicide Triclabendazole to Treat Infections Caused by Staphylococcus spp. and Vancomycin-Resistant Enterococci. Microorganisms 2021, 9, 1697. https://doi.org/10.3390/microorganisms9081697
Pi H, Ogunniyi AD, Savaliya B, Nguyen HT, Page SW, Lacey E, Venter H, Trott DJ. Repurposing of the Fasciolicide Triclabendazole to Treat Infections Caused by Staphylococcus spp. and Vancomycin-Resistant Enterococci. Microorganisms. 2021; 9(8):1697. https://doi.org/10.3390/microorganisms9081697
Chicago/Turabian StylePi, Hongfei, Abiodun D. Ogunniyi, Bhumi Savaliya, Hang Thi Nguyen, Stephen W. Page, Ernest Lacey, Henrietta Venter, and Darren J. Trott. 2021. "Repurposing of the Fasciolicide Triclabendazole to Treat Infections Caused by Staphylococcus spp. and Vancomycin-Resistant Enterococci" Microorganisms 9, no. 8: 1697. https://doi.org/10.3390/microorganisms9081697
APA StylePi, H., Ogunniyi, A. D., Savaliya, B., Nguyen, H. T., Page, S. W., Lacey, E., Venter, H., & Trott, D. J. (2021). Repurposing of the Fasciolicide Triclabendazole to Treat Infections Caused by Staphylococcus spp. and Vancomycin-Resistant Enterococci. Microorganisms, 9(8), 1697. https://doi.org/10.3390/microorganisms9081697