
Clary Sage Cultivation and Mycorrhizal Inoculation Influence the Rhizosphere Fungal Community of an Aged Trace-Element Polluted Soil




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Site
2.2. Biological Material
2.3. Experimental Design
2.4. Soil and Plant Roots Sampling Procedure
2.5. DNA Extractions
2.5.1. Soil Samples
2.5.2. Plant Root Samples
2.6. PCR and Sequencing
2.6.1. Fungal ITS
2.6.2. 18S rRNA Gene of AMF
2.6.3. Illumina MiSeq Sequencing
2.7. Bioinformatic Processing
2.8. Nucleotide Sequence Accession Number
2.9. Statistical Analysis
3. Results
3.1. Raw Sequences Bioinformatic Processing
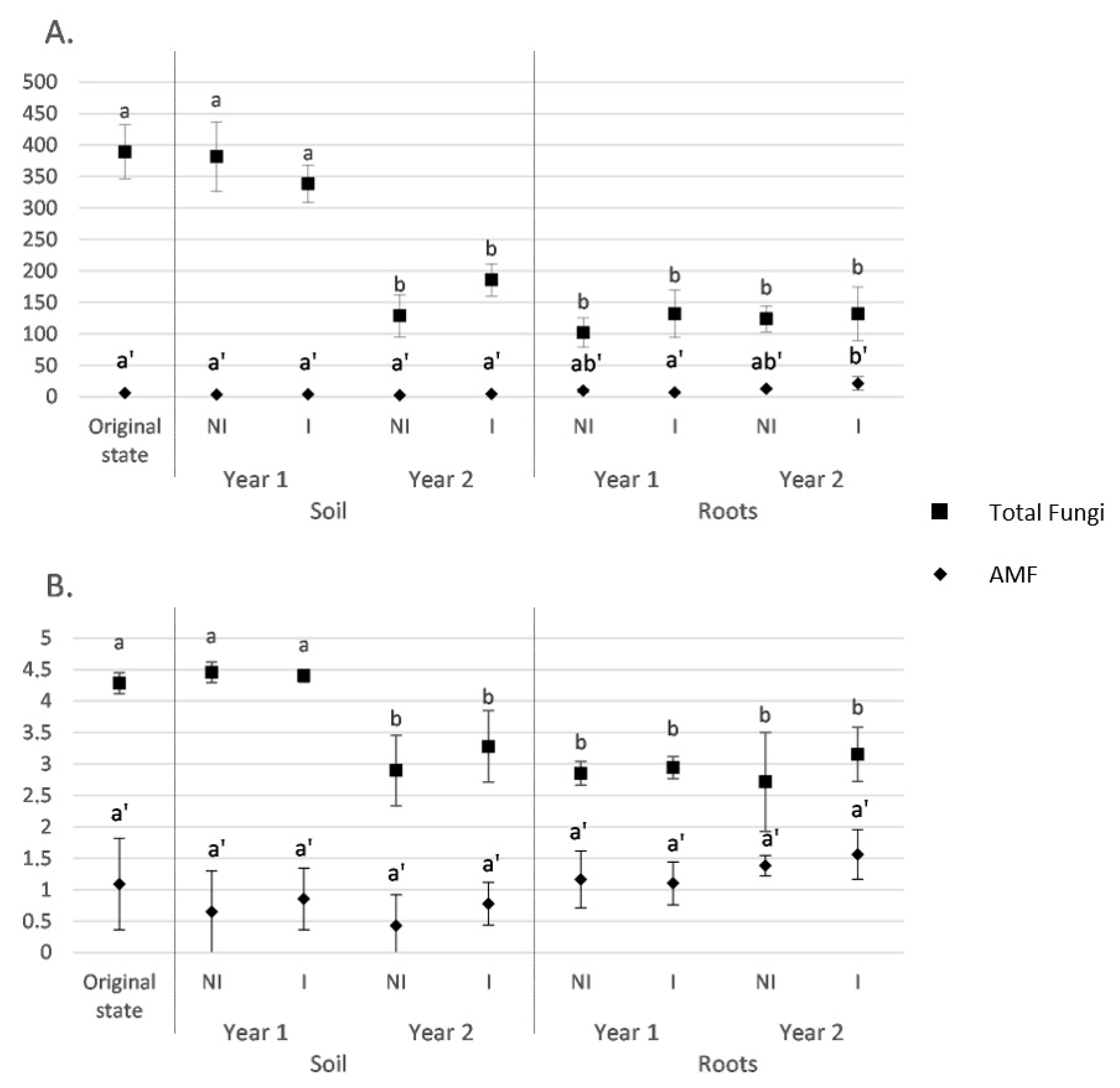
3.2. Fungal and AMF α-Diversity in Root and Soil Biotopes
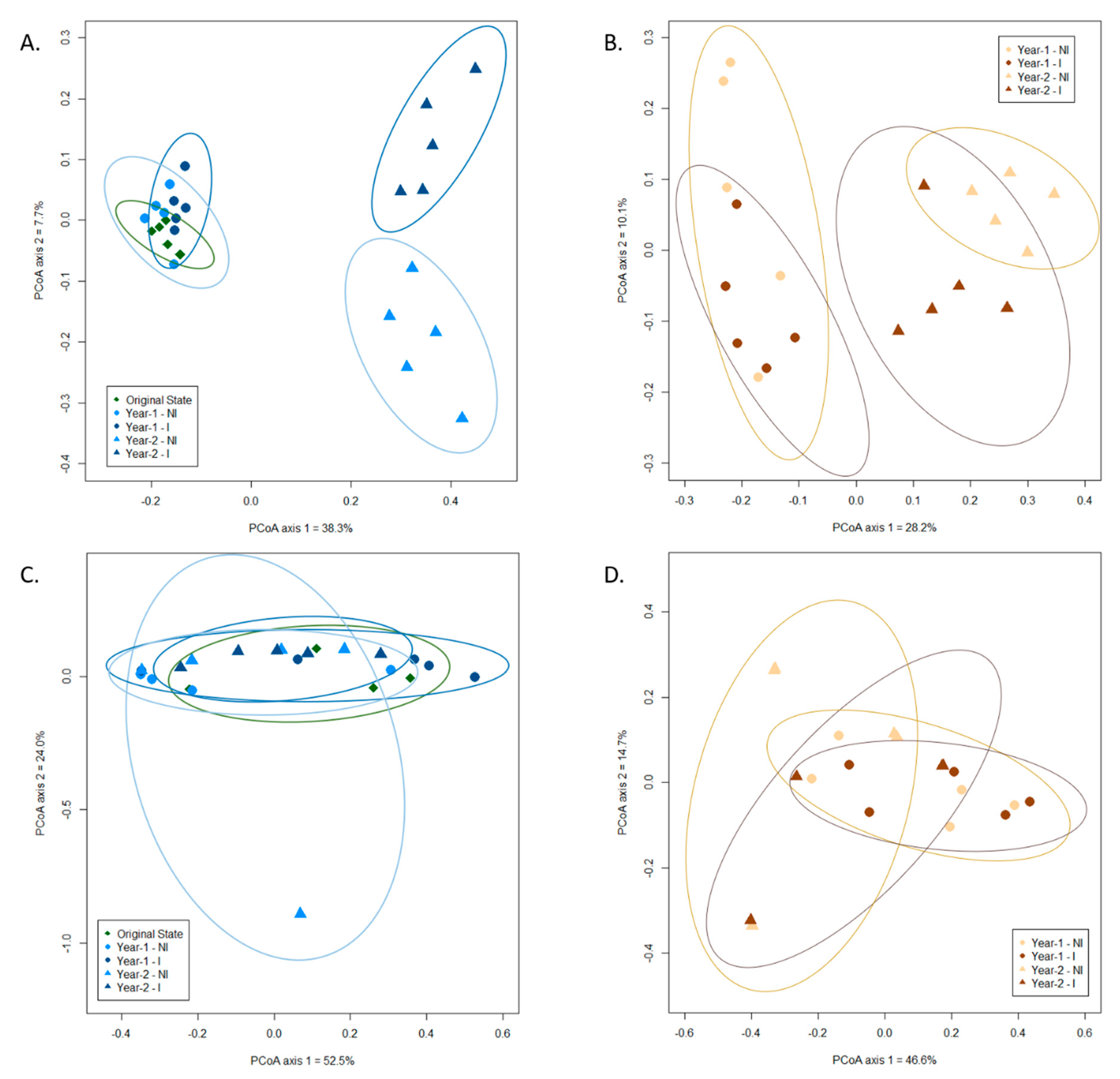
3.3. Influence of Mycorrhizal Inoculation and Cultivation Duration on the Fungal Community Composition
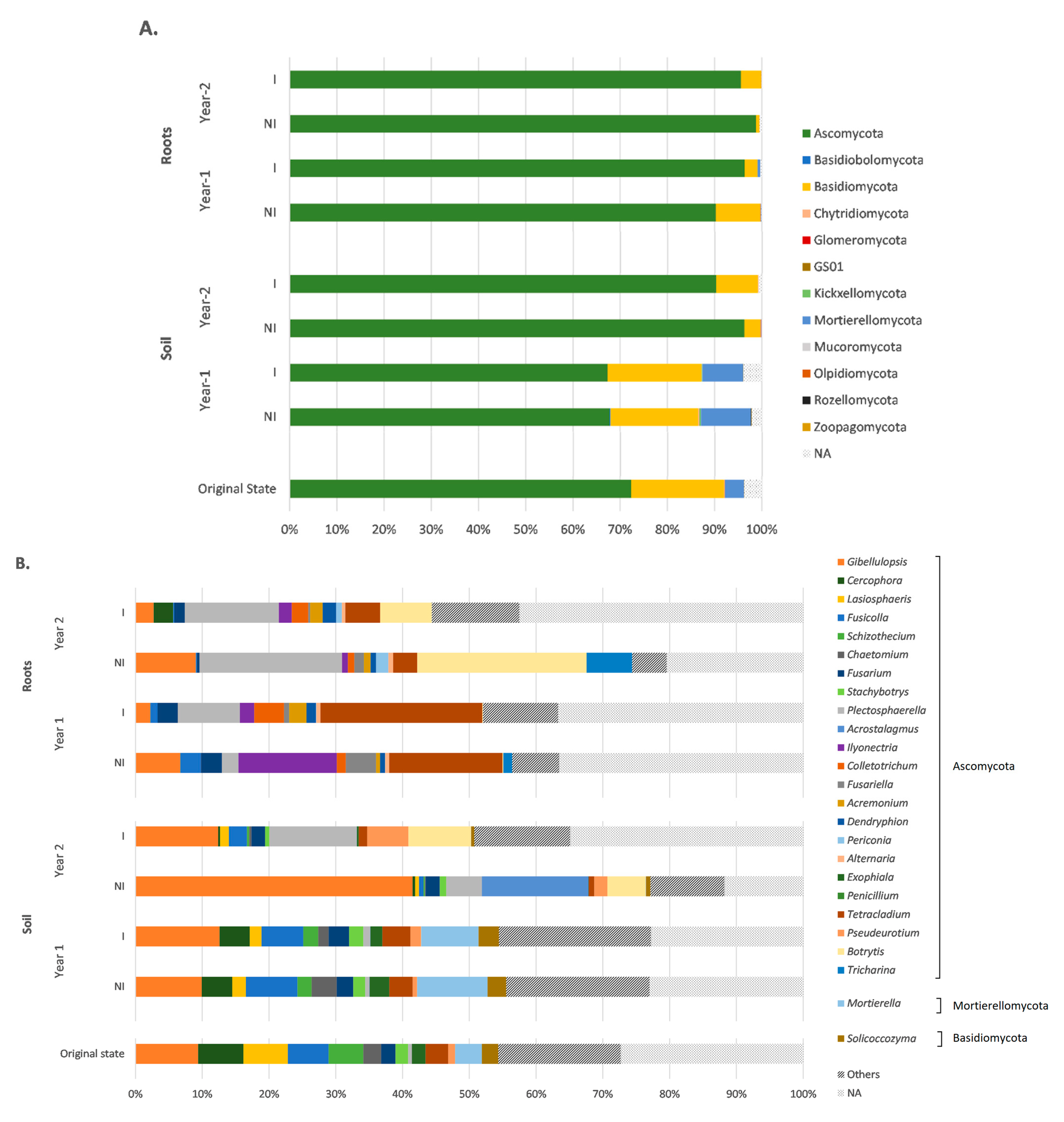
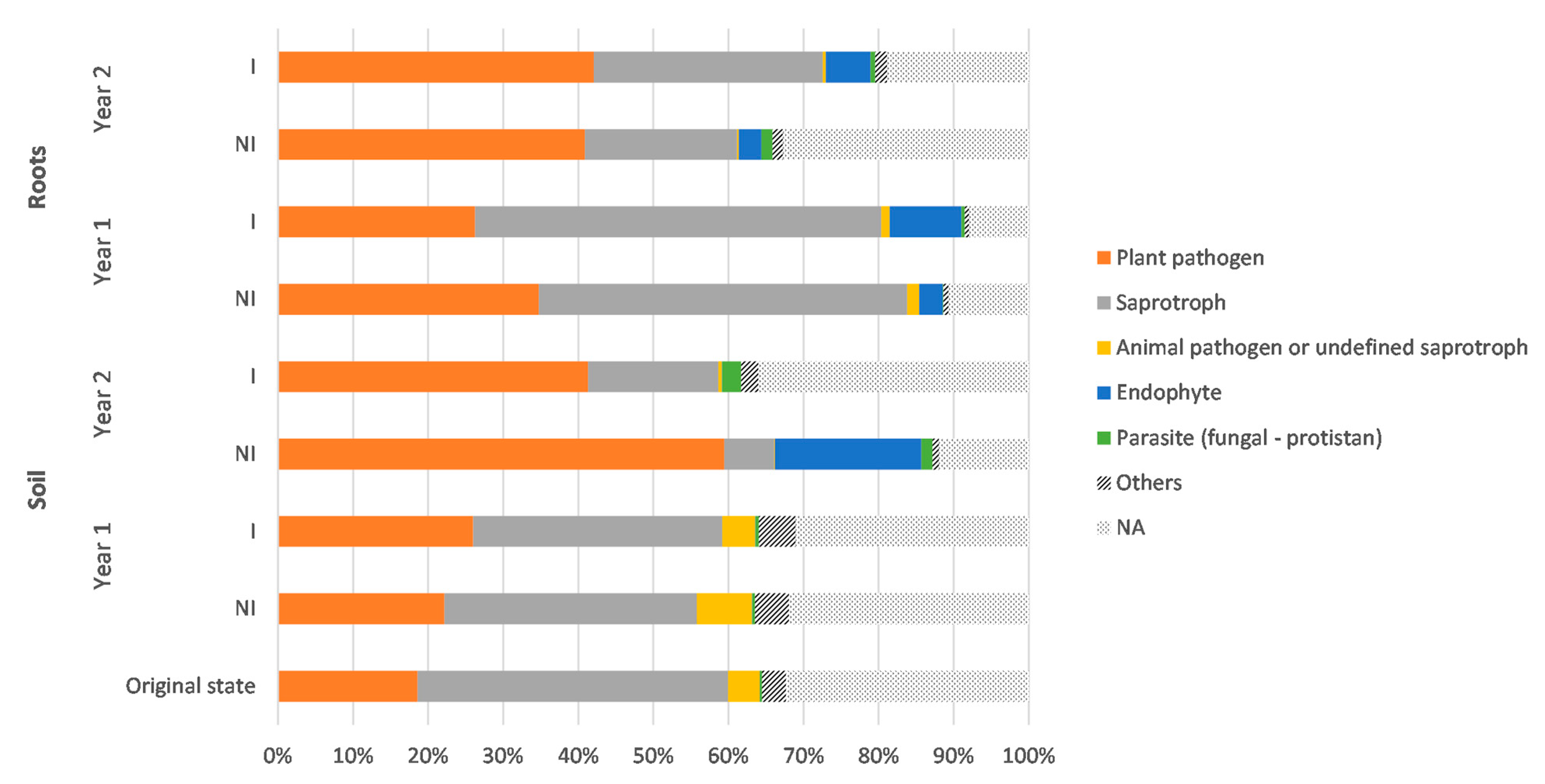
3.4. Taxonomic Variations in the Fungal Communities in Response to Mycorrhizal Inoculation and Cultivation Time
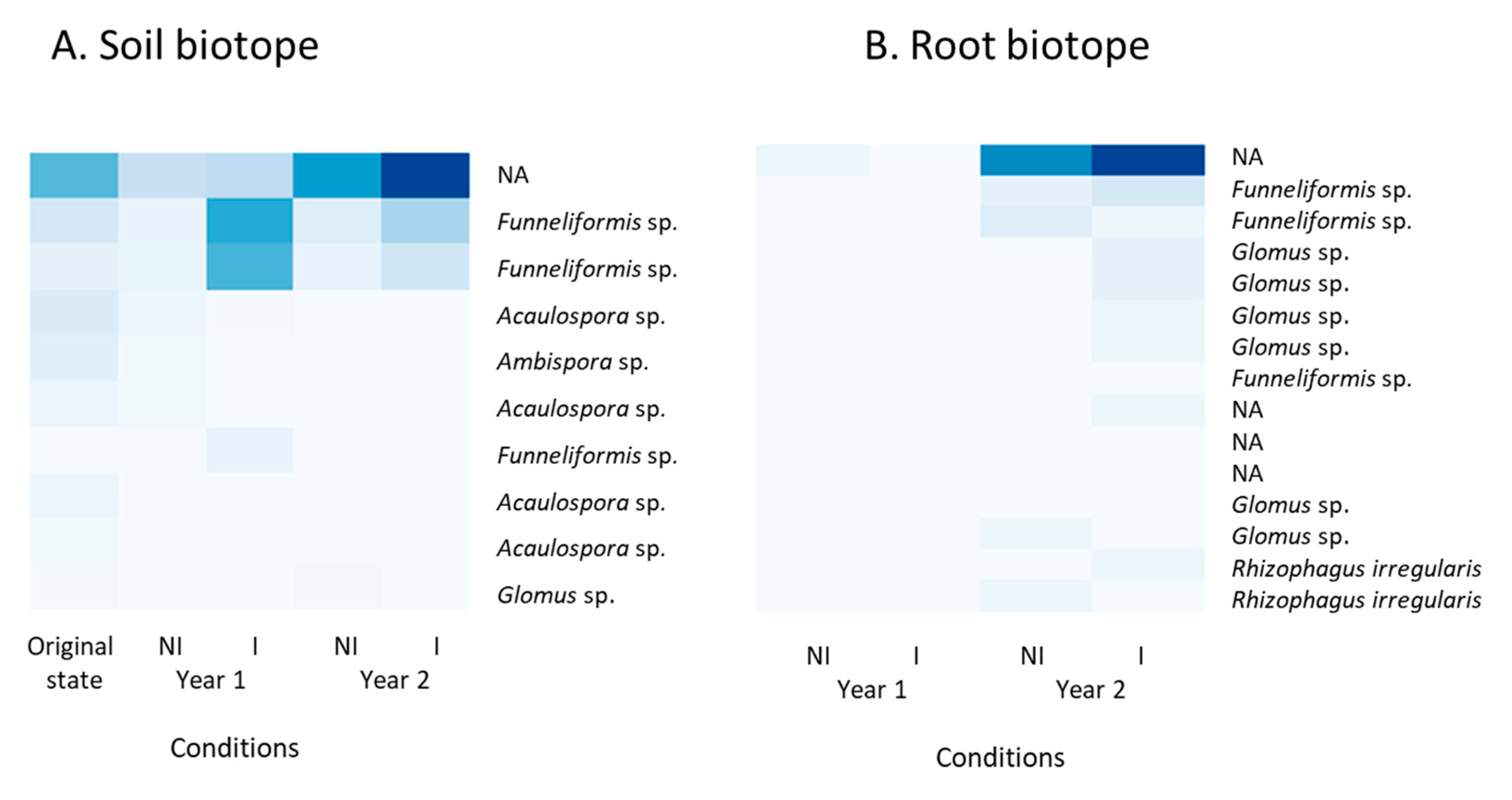
3.5. Taxonomic Identification of ASVs Belonging to the AMF 18S rRNA Gene Data Set
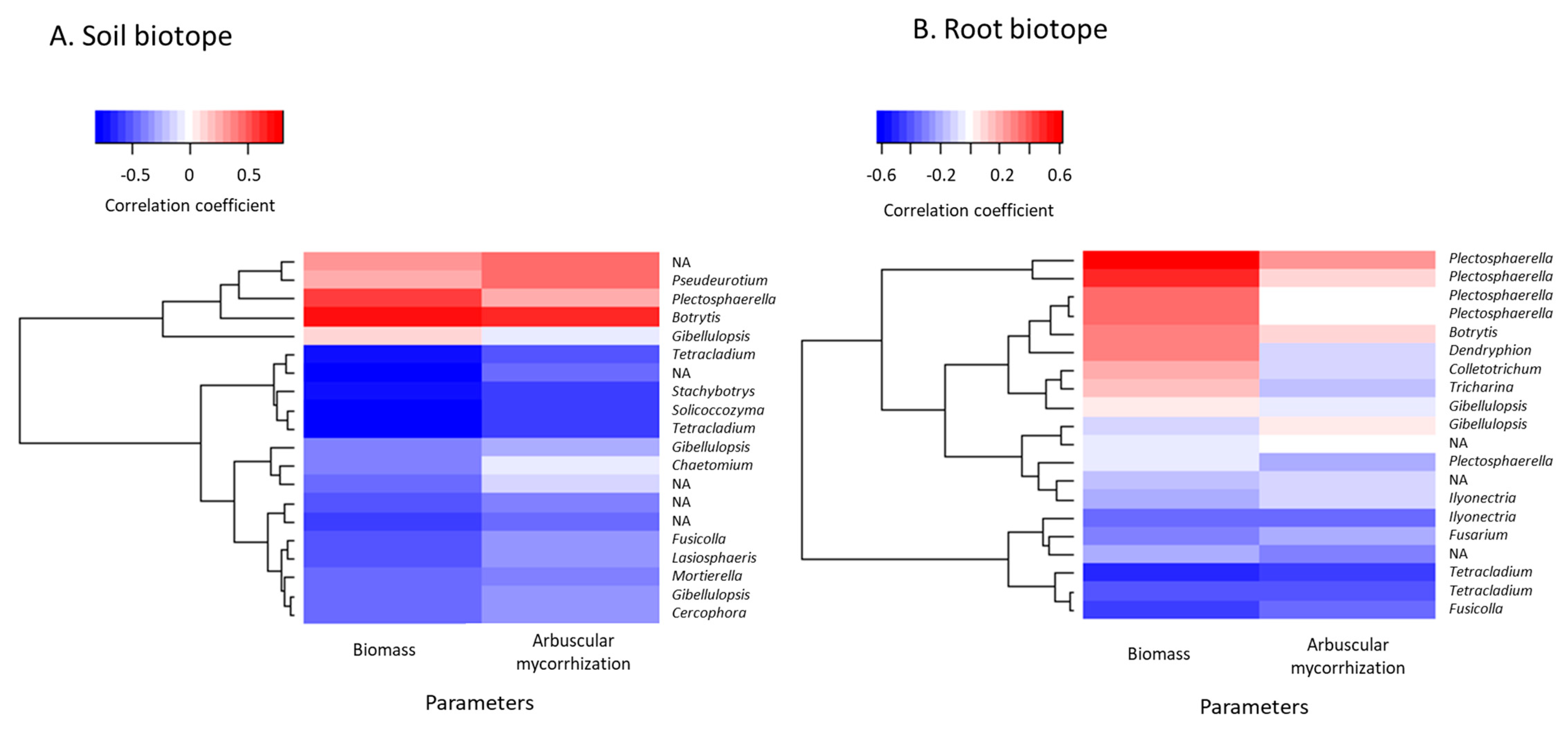
3.6. Prediction of Functional Assignments of ASVs
4. Discussion
4.1. Ascomycota Phylum Dominates the Fungal Community in an Aged TE-Polluted Soil before Clary Sage Cultivation
4.2. Clary Sage Significantly Shaped the Rhizosphere Fungal Community over its Life Cycle
4.3. Mycorrhizal Inoculation Shaped the Rhizosphere Fungal Community and Could Favor the Establishment of the Mycorrhizal Symbiosis
4.4. The AMF Community Changed over Time under Clary Sage Cultivation, but was Not Altered by Exogenous Mycorrhizal Inoculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ding, Z.; Wu, J.; You, A.; Huang, B.; Cao, C. Effects of heavy metals on soil microbial community structure and diversity in the rice (Oryza sativa L. subsp. Japonica, Food Crops Institute of Jiangsu Academy of Agricultural Sciences) rhizosphere. Soil Sci. Plant Nutr. 2017, 63, 75–83. [Google Scholar] [CrossRef]
- Adamovic, D.; Djalovic, I.; Mrkovacki, N. Microbial abundance in rhizosphere of medicinal and aromatic plant species in conventional and organic growing systems. Ratar. Povrt. 2015, 52, 1–6. [Google Scholar] [CrossRef]
- Klaubauf, S.; Inselsbacher, E.; Zechmeister-Boltenstern, S.; Wanek, W.; Gottsberger, R.; Strauss, J.; Gorfer, M. Molecular diversity of fungal communities in agricultural soils from Lower Austria. Fungal Divers. 2010, 44, 65–75. [Google Scholar] [CrossRef]
- Bodelier, P.L.E. Toward understanding, managing, and protecting microbial ecosystems. Front. Microbiol. 2011, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Wang, R.; Fan, X.; Chai, B. A comparative study of fungal community structure, diversity and richness between the soil and the phyllosphere of native grass species in a copper tailings dam in Shanxi Province, China. Appl. Sci. 2018, 8, 1297. [Google Scholar] [CrossRef]
- Lin, Y.; Xiao, W.; Ye, Y.; Wu, C.; Hu, Y.; Shi, H. Adaptation of soil fungi to heavy metal contamination in paddy fields—A case study in eastern China. Environ. Sci. Pollut. Res. 2020, 27, 27819–27830. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Liu, J.; Yang, T.; Wang, P.; Zhang, S.; Jia, P.; Liao, B.; Shu, W.; Li, J. Contrasting soil fungal communities at different habitats in a revegetated copper mine wasteland. Soil Ecol. Lett. 2020, 2, 8–19. [Google Scholar] [CrossRef]
- Smith, S.; Read, D. Mycorrhizal Symbiosis; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 978-0-12370-526-6. [Google Scholar]
- Janoušková, M.; Krak, K.; Vosátka, M.; Püschel, D.; Štorchová, H. Inoculation effects on root-colonizing arbuscular mycorrhizal fungal communities spread beyond directly inoculated plants. PLoS ONE 2017, 12, e0181525. [Google Scholar] [CrossRef]
- Dagher, D.J.; De La Providencia, I.E.; Pitre, F.E.; St-Arnaud, M.; Hijri, M. Arbuscular mycorrhizal fungal assemblages significantly shifted upon bacterial inoculation in non-contaminated and petroleum-contaminated environments. Microorganisms 2020, 8, 602. [Google Scholar] [CrossRef]
- Gong, Y.; Zhao, D.; Wang, Q. An overview of field-scale studies on remediation of soil contaminated with heavy metals and metalloids: Technical progress over the last decade. Water Res. 2018, 147, 440–460. [Google Scholar] [CrossRef]
- Ferrol, N.; Tamayo, E.; Vargas, P. The heavy metal paradox in arbuscular mycorrhizas: From mechanisms to biotechnological applications. J. Exp. Bot. 2016, 67, 6253–6565. [Google Scholar] [CrossRef]
- Op De Beeck, M.; Lievens, B.; Busschaert, P.; Rineau, F.; Smits, M.; Vangronsveld, J.; Colpaert, J.V. Impact of metal pollution on fungal diversity and community structures. Environ. Microbiol. 2015, 17, 2035–2047. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ye, Y.; Hu, Y.; Shi, H. The variation in microbial community structure under different heavy metal contamination levels in paddy soils. Ecotoxicol. Environ. Saf. 2019, 180, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Järup, L. Hazards of heavy metal contamination. Br. Med. Bull. 2003, 68, 167–182. [Google Scholar] [CrossRef]
- Chen, J.; He, F.; Zhang, X.; Sun, X.; Zheng, J.; Zheng, J. Heavy metal pollution decreases microbial abundance, diversity and activity within particle-size fractions of a paddy soil. FEMS Microbiol. Ecol. 2014, 87, 164–181. [Google Scholar] [CrossRef]
- Bourceret, A.; Cébron, A.; Tisserant, E.; Poupin, P.; Bauda, P.; Beguiristain, T.; Leyval, C. The Bacterial and Fungal Diversity of an Aged PAH- and Heavy Metal-Contaminated Soil is Affected by Plant Cover and Edaphic Parameters. Microb. Ecol. 2016, 71, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Burges, A.; Alkorta, I.; Epelde, L.; Garbisu, C. From phytoremediation of soil contaminants to phytomanagement of ecosystem services in metal contaminated sites. Int. J. Phytoremediat. 2018, 20, 384–397. [Google Scholar] [CrossRef]
- Mench, M.J.; Dellise, M.; Bes, C.M.; Marchand, L.; Kolbas, A.; Le Coustumer, P.; Oustrière, N. Phytomanagement and remediation of cu-contaminated soils by high yielding crops at a former wood preservation site: Sunflower biomass and ionome. Front. Ecol. Evol. 2018, 6. [Google Scholar] [CrossRef]
- Cundy, A.B.; Bardos, R.P.; Puschenreiter, M.; Mench, M.; Bert, V.; Friesl-Hanl, W.; Müller, I.; Li, X.N.; Weyens, N.; Witters, N.; et al. Brownfields to green fields: Realising wider benefits from practical contaminant phytomanagement strategies. J. Environ. Manag. 2016, 184, 67–77. [Google Scholar] [CrossRef]
- Evangelou, M.W.H.; Papazoglou, E.G.; Robinson, B.H.; Schulin, R. Phytomanagement: Phytoremediation and the Production of Biomass for Economic Revenue on Contaminated Land. In Phytoremediation: Management of Environmental Contaminants; Springer: Berlin, Germany, 2015; Volume 1, ISBN 978-3-31910-395-2. [Google Scholar]
- Pandey, V.C.; Bajpai, O.; Singh, N. Energy crops in sustainable phytoremediation. Renew. Sustain. Energy Rev. 2016, 54, 58–73. [Google Scholar] [CrossRef]
- Bauddh, K.; Singh, B.; Korstad, J. Phytoremediation Potential of Bioenergy Plants; Springer: Berlin, Germany, 2017; ISBN 978-9-81103-084-0. [Google Scholar]
- Tibaldi, G.; Fontana, E.; Nicola, S. Cultivation practices do not change the Salvia sclarea L. essential oil but drying process does. J. Food Agric. Environ. 2010, 8, 790–794. [Google Scholar]
- Grigoriadou, K.; Trikka, F.A.; Tsoktouridis, G.; Krigas, N.; Sarropoulou, V.; Papanastasi, K.; Maloupa, E.; Makris, A.M. Μicropropagation and cultivation of Salvia sclarea for essential oil and sclareol production in northern Greece. In Vitro Cell. Dev. Biol. Plant 2020, 56, 51–59. [Google Scholar] [CrossRef]
- Lydakis-Simantiris, N.; Fabian, M.; Skoula, M. Cultivation of medicinal and aromatic plants in heavy metal-contaminated soils. Glob. Nest J. 2016, 18, 630–642. [Google Scholar] [CrossRef]
- Angelova, V.R.; Ivanova, R.; Todorov, G.M.; Ivanov, K.I. Potential of Salvia sclarea L. for Phytoremediation of Soils Contaminated with Heavy Metals. Agric. Biosyst. Eng. 2016, 10, 780–790. [Google Scholar]
- Zutic, I.; Nitzan, N.; Chaimovitsh, D.; Schechter, A.; Dudai, N. Geographical location is a key component to effective breeding of clary sage (Salvia sclarea) for essential oil composition. Isr. J. Plant Sci. 2016, 63, 134–141. [Google Scholar] [CrossRef]
- Aćimović, M.; Kiprovski, B.; Rat, M.; Sikora, V.; Popović, V.; Koren, A.; Brdar-Jokanović, M. Salvia sclarea: Chemical composition and biological activity. J. Agron. Technol. Eng. Manag. 2018, 1, 18–28. [Google Scholar]
- Dagher, D.J.; Pitre, F.E.; Hijri, M. Ectomycorrhizal Fungal Inoculation of Sphaerosporella brunnea Significantly Increased Stem Biomass of Salix miyabeana and Decreased Lead, Tin, and Zinc, Soil Concentrations during the Phytoremediation of an Industrial Landfill. J. Fungi 2020, 6, 87. [Google Scholar] [CrossRef]
- Antoniadis, V.; Levizou, E.; Shaheen, S.M.; Ok, Y.S.; Sebastian, A.; Baum, C.; Prasad, M.N.V.; Wenzel, W.W.; Rinklebe, J. Trace elements in the soil-plant interface: Phytoavailability, translocation, and phytoremediation—A review. Earth Sci. Rev. 2017, 171, 621–645. [Google Scholar] [CrossRef]
- Akyol, T.Y.; Niwa, R.; Hirakawa, H.; Maruyama, H.; Sato, T.; Suzuki, T.; Fukunaga, A.; Sato, T.; Yoshida, S.; Tawaraya, K.; et al. Impact of introduction of arbuscular mycorrhizal fungi on the root microbial community in agricultural fields. Microbes Environ. 2019, 34, 23–32. [Google Scholar] [CrossRef]
- Lioussanne, L.; Perreault, F.; Jolicoeur, M.; St-Arnaud, M. The bacterial community of tomato rhizosphere is modified by inoculation with arbuscular mycorrhizal fungi but unaffected by soil enrichment with mycorrhizal root exudates or inoculation with Phytophthora nicotianae. Soil Biol. Biochem. 2010, 42, 473–483. [Google Scholar] [CrossRef]
- Koch, A.M.; Antunes, P.M.; Barto, E.K.; Cipollini, D.; Mummey, D.L.; Klironomos, J.N. The effects of arbuscular mycorrhizal (AM) fungal and garlic mustard introductions on native AM fungal diversity. Biol. Invasions 2011, 13, 1627–1639. [Google Scholar] [CrossRef]
- Renaut, S.; Daoud, R.; Masse, J.; Vialle, A.; Hijri, M. Inoculation with Rhizophagus irregularis does not alter arbuscular mycorrhizal fungal community structure within the roots of corn, wheat, and soybean crops. Microorganisms 2020, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Janoušková, M.; Krak, K.; Wagg, C.; Štorchová, H.; Caklová, P.; Vosátka, M. Effects of inoculum additions in the presence of a preestablished arbuscular mycorrhizal fungal community. Appl. Environ. Microbiol. 2013, 79, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, D.; Mhamdi, R. Microbial inoculants and their impact on soil microbial communities: A review. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Iffis, B.; St-Arnaud, M.; Hijri, M. Petroleum hydrocarbon contamination, plant identity and arbuscular mycorrhizal fungal (AMF) community determine assemblages of the AMF spore-associated microbes. Environ. Microbiol. 2016, 18, 2689–2704. [Google Scholar] [CrossRef] [PubMed]
- Dagher, D.J.; de la Providencia, I.E.; Pitre, F.E.; St-Arnaud, M.; Hijri, M. Plant Identity Shaped Rhizospheric Microbial Communities More Strongly Than Bacterial Bioaugmentation in Petroleum Hydrocarbon-Polluted Sediments. Front. Microbiol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Labidi, S.; Fontaine, J.; Laruelle, F.; Tisserant, B.; Dalpé, Y.; Grandmougin-Ferjani, A.; Douay, F.; Lounès-Hadj Sahraoui, A. Fly ash-aided phytostabilisation of highly trace element polluted topsoils improves the telluric fungal biomass: A long-term field experiment. Appl. Soil Ecol. 2015, 85, 69–75. [Google Scholar] [CrossRef]
- Sterckeman, T.; Douay, F.; Proix, N.; Fourrier, H.; Perdrix, E. Assessment of the contamination of cultivated soils by eighteen trace elements around smelters in the north of France. Appl. Soil Ecol. 2002, 85, 173–194. [Google Scholar] [CrossRef]
- Sterckeman, T.; Douay, F.; Baize, D.; Fourrier, H.; Proix, N.; Schvartz, C. Référentiel pédo-géochimique du Nord-Pas de Calais: Méthode et principaux résultats. Etude Gest. Sols 2007, 14, 153–168. [Google Scholar]
- Raveau, R.; Fontaine, J.; Hijri, M.; Lounès-Hadj Sahraoui, A. The Aromatic Plant Clary Sage Shaped Bacterial Communities in the Roots and in the Trace Element-Contaminated Soil More Than Mycorrhizal Inoculation—A Two-Year Monitoring Field Trial. Front. Microbiol. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Raveau, R.; Fontaine, J.; Bert, V.; Perlein, A.; Tisserant, B.; Ferrant, P.; Lounès-Hadj Sahraoui, A. In situ cultivation of aromatic plant species for the phytomanagement of an aged-trace element polluted soil: Plant biomass improvement options and techno-economic assessment of the essential oil production channel. Sci. Total Environ. 2021, 147944. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Yi, K.; Fang, K.; Gao, H.; Dai, W.; Cao, L. Microbial Community Structures and Important Associations Between Soil Nutrients and the Responses of Specific Taxa to Rice-Frog Cultivation. Front. Microbiol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liu, G.; Xue, S. Interaction between plant competition and rhizospheric bacterial community influence secondary succession of abandoned farmland on the loess plateau of China. Front. Plant Sci. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abu-Romman, S. Comparison of methods for isolating high quality dna from sage (Salvia officinalis). J. Med. Plants Res. 2011, 5, 938–941. [Google Scholar]
- Aleksić, J.M.; Stojanović, D.; Banović, B.; Jančić, R. A simple and efficient DNA isolation method for Salvia officinalis. Biochem. Genet. 2012, 50, 881–892. [Google Scholar] [CrossRef]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Young, J.P.W. Improved PCR primers for the detection and identification of arbuscular mycorrhizal fungi. FEMS Microbiol. Ecol. 2008, 65, 339–349. [Google Scholar] [CrossRef]
- Stefani, F.; Bencherif, K.; Sabourin, S.; Lounès-Hadj Sahraoui, A.; Banchini, C.; Séguin, S.; Dalpé, Y. Taxonomic assignment of arbuscular mycorrhizal fungi in an 18S metagenomic dataset: A case study with saltcedar (Tamarix aphylla). Mycorrhiza 2020, 30, 243–255. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Callahan, B. Silva taxonomic training data formatted for DADA2 (Silva version 132). Zenodo 2018. [Google Scholar] [CrossRef]
- Krüger, M.; Krüger, C.; Walker, C.; Stockinger, H.; Schüßler, A. Phylogenetic reference data for systematics and phylotaxonomy of arbuscular mycorrhizal fungi from phylum to species level. New Phytol. 2012, 193, 970–984. [Google Scholar] [CrossRef]
- Lassmann, T.; Frings, O.; Sonnhammer, E.L.L. Kalign2: High-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 2009, 37, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Shi, Y.; Qiu, L.; Guo, L.; Man, J.; Shang, B.; Pu, R.; Ou, X.; Dai, C.; Liu, P.; Yang, Y.; et al. K Fertilizers Reduce the Accumulation of Cd in Panax notoginseng (Burk.) F.H. by Improving the Quality of the Microbial Community. Front. Plant Sci. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Torres-Cruz, T.J.; Hesse, C.; Kuske, C.R.; Porras-Alfaro, A. Presence and distribution of heavy metal tolerant fungi in surface soils of a temperate pine forest. Appl. Soil Ecol. 2018, 131, 66–74. [Google Scholar] [CrossRef]
- Misra, P.; Maji, D.; Awasthi, A.; Pandey, S.S.; Yadav, A.; Pandey, A.; Saikia, D.; Babu, C.S.V.; Kalra, A. Vulnerability of Soil Microbiome to Monocropping of Medicinal and Aromatic Plants and Its Restoration Through Intercropping and Organic Amendments. Front. Microbiol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Blackwood, C.B.; Waldrop, M.P.; Zak, D.R.; Sinsabaugh, R.L. Molecular analysis of fungal communities and laccase genes in decomposing litter reveals differences among forest types but no impact of nitrogen deposition. Environ. Microbiol. 2007, 9, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.B.; Wu, C.; Zhang, C.; Li, H.; Lipka, U.; Polle, A. The role of ectomycorrhizas in heavy metal stress tolerance of host plants. Environ. Exp. Bot. 2014, 108, 47–62. [Google Scholar] [CrossRef]
- Kalsotra, T.; Khullar, S.; Agnihotri, R.; Reddy, M.S. Metal induction of two metallothionein genes in the ectomycorrhizal fungus suillus himalayensis and their role in metal tolerance. Microbiology 2018, 164, 868–876. [Google Scholar] [CrossRef]
- Shine, A.M.; Shakya, V.P.; Idnurm, A. Phytochelatin synthase is required for tolerating metal toxicity in a basidiomycete yeast and is a conserved factor involved in metal homeostasis in fungi. Fungal Biol. Biotechnol. 2015, 2, 1–13. [Google Scholar] [CrossRef]
- Sudhakara Reddy, M.; Prasanna, L.; Marmeisse, R.; Fraissinet-Tachet, L. Differential expression of metallothioneins in response to heavy metals and their involvement in metal tolerance in the symbiotic basidiomycete Laccaria bicolor. Microbiology 2014, 160, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Foulon, J.; Zappelini, C.; Durand, A.; Valot, B.; Blaudez, D.; Chalot, M. Impact of poplar-based phytomanagement on soil properties and microbial communities in a metal-contaminated site. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed]
- Lopes Leal, P.; Varón-López, M.; Gonçalves de Oliveira Prado, I.; Valentim dos Santos, J.; Fonsêca Sousa Soares, C.R.; Siqueira, J.O.; de Souza Moreira, F.M. Enrichment of arbuscular mycorrhizal fungi in a contaminated soil after rehabilitation. Braz. J. Microbiol. 2016, 47, 853–862. [Google Scholar] [CrossRef]
- Yang, Y.; Song, Y.; Scheller, H.V.; Ghosh, A.; Ban, Y.; Chen, H.; Tang, M. Community structure of arbuscular mycorrhizal fungi associated with Robinia pseudoacacia in uncontaminated and heavy metal contaminated soils. Soil Biol. Biochem. 2015, 86, 146–158. [Google Scholar] [CrossRef]
- Goldmann, K.; Schröter, K.; Pena, R.; Schöning, I.; Schrumpf, M.; Buscot, F.; Polle, A.; Wubet, T. Divergent habitat filtering of root and soil fungal communities in temperate beech forests. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, A.; Hernández-Restrepo, M.; Crous, P.W. New plectosphaerellaceous species from Dutch garden soil. Mycol. Prog. 2019, 18, 1135–1154. [Google Scholar] [CrossRef]
- Carlucci, A.; Raimondo, M.L.; Santos, J.; Phillips, A.J.L. Plectosphaerella species associated with root and collar rots of horticultural crops in southern Italy. Pers. Mol. Phylogeny Evol. Fungi 2012, 28, 34–48. [Google Scholar] [CrossRef]
- Wang, Z.; Li, T.; Wen, X.; Liu, Y.; Han, J.; Liao, Y.; DeBruyn, J.M. Fungal communities in rhizosphere soil under conservation tillage shift in response to plant growth. Front. Microbiol. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Na, X.; Ma, C.; Ma, S.; Ma, X.; Zhu, X.; Xu, P.; Zhu, H.; Cao, X.; Liang, W. Monocropping decouples plant–bacteria interaction and strengthens phytopathogenic fungi colonization in the rhizosphere of a perennial plant species. Plant Soil 2019, 445, 549–564. [Google Scholar] [CrossRef]
- Qu, Z.; Liu, B.; Ma, Y.; Sun, H. Differences in bacterial community structure and potential functions among Eucalyptus plantations with different ages and species of trees. Appl. Soil Ecol. 2020, 149, 103515. [Google Scholar] [CrossRef]
- Wei, X.; Wang, X.; Cao, P.; Gao, Z.; Chen, A.J.; Han, J. Microbial Community Changes in the Rhizosphere Soil of Healthy and Rusty Panax ginseng and Discovery of Pivotal Fungal Genera Associated with Rusty Roots. Biomed Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Puškárová, A.; Bučková, M.; Kraková, L.; Pangallo, D.; Kozics, K. The antibacterial and antifungal activity of six essential oils and their cyto/genotoxicity to human HEL 12469 cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pitarokili, D.; Couladis, M.; Petsikos-Panayotarou, N.; Tzakou, O. Composition and antifungal activity on soil-borne pathogens of the essential oil of Salvia sclarea from Greece. J. Agric. Food Chem. 2002, 50, 6688–6691. [Google Scholar] [CrossRef]
- Li, X.; Panke-Buisse, K.; Yao, X.; Coleman-Derr, D.; Ding, C.; Wang, X.; Ruan, H. Peanut plant growth was altered by monocropping-associated microbial enrichment of rhizosphere microbiome. Plant Soil 2020, 446, 655–669. [Google Scholar] [CrossRef]
- Li, W.H.; Liu, Q.Z. Changes in fungal community and diversity in strawberry rhizosphere soil after 12 years in the greenhouse. J. Integr. Agric. 2019, 18, 677–687. [Google Scholar] [CrossRef]
- Iffis, B.; St-Arnaud, M.; Hijri, M. Bacteria associated with arbuscular mycorrhizal fungi within roots of plants growing in a soil highly contaminated with aliphatic and aromatic petroleum hydrocarbons. FEMS Microbiol. Lett. 2014, 358, 44–54. [Google Scholar] [CrossRef]
- Hijri, M.; Redecker, D.; MacDonald-Comber Petetot, J.A.; Voigt, K.; Wöstemeyer, J.; Sanders, I.R. Identification and isolation of two ascomycete fungi from spores of the arbuscular mycorrhizal fungus Scutellospora castanea. Appl. Environ. Microbiol. 2002, 68, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Zappelini, C.; Karimi, B.; Foulon, J.; Lacercat-Didier, L.; Maillard, F.; Valot, B.; Blaudez, D.; Cazaux, D.; Gilbert, D.; Yergeau, E.; et al. Diversity and complexity of microbial communities from a chlor-alkali tailings dump. Soil Biol. Biochem. 2015. [Google Scholar] [CrossRef]
- Zhao, M.; Sun, B.; Wu, L.; Wang, F.; Wen, C.; Wang, M.; Liang, Y.; Hale, L.; Zhou, J.; Yang, Y. Dissimilar responses of fungal and bacterial communities to soil transplantation simulating abrupt climate changes. Mol. Ecol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Zhang, Y.; Zou, H. Plant growth and their root development after inoculation of arbuscular mycorrhizal fungi in coal mine subsided areas. Int. J. Coal Sci. Technol. 2018, 5, 47–53. [Google Scholar] [CrossRef]
- Griffiths, B.S.; Philippot, L. Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol. Rev. 2013, 37, 112–129. [Google Scholar] [CrossRef]
- Sharma, S.; Kumar, R. Effect of nitrogen on growth, biomass and oil composition of clary sage (Salvia sclarea Linn.) under mid hills of north western Himalayas. Indian J. Nat. Prod. Resour. 2012, 3, 79–83. [Google Scholar]
- Abbaszadeh, B.; Safikhani, F.; Layeghhaghighi, M. Effects of Irrigation Interval and Nitrogen Amount on Different Clary Sage (Salvia sclarea L.) Characters in Karaj. J. Med. Plants By-Prod. JMPB 2017, 6, 139–144. [Google Scholar]
- Denoroy, P.; Dubrulle, P.; Villette, C.; Colomb, B. REGIFERT, Interpréter les Résultats des Analyses de Terre; Quae, E., Ed.; INRA Editions: Paris, France, 2004; ISBN 978-2-73801-168-8. [Google Scholar]
- Karandashov, V.; Bucher, M. Symbiotic phosphate transport in arbuscular mycorrhizas. Trends Plant Sci. 2005, 10, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Voß, S.; Betz, R.; Heidt, S.; Corradi, N.; Requena, N. RiCRN1, a crinkler effector from the arbuscular mycorrhizal fungus rhizophagus irregularis, functions in arbuscule development. Front. Microbiol. 2018, 9, 1–18. [Google Scholar] [CrossRef]
- Paszkowski, U.; Kroken, S.; Roux, C.; Briggs, S.P. Rice phosphate transporters include an evolutionarily divergent gene specifically activated in arbuscular mycorrhizal symbiosis. Proc. Natl. Acad. Sci. USA 2002, 99, 13324–13329. [Google Scholar] [CrossRef]
- Hart, M.M.; Antunes, P.M.; Chaudhary, V.B.; Abbott, L.K. Fungal inoculants in the field: Is the reward greater than the risk? Funct. Ecol. 2017, 32, 126–135. [Google Scholar] [CrossRef]
- Saks, Ü.; Davison, J.; Öpik, M.; Vasar, M.; Moora, M.; Zobel, M. Root-colonizing and soil-borne communities of arbuscular mycorrhizal fungi in a temperate forest understorey. Botany 2014, 92, 277–285. [Google Scholar] [CrossRef]
- Varela-Cervero, S.; Vasar, M.; Davison, J.; Barea, J.M.; Öpik, M.; Azcón-Aguilar, C. The composition of arbuscular mycorrhizal fungal communities differs among the roots, spores and extraradical mycelia associated with five Mediterranean plant species. Environ. Microbiol. 2015, 17, 2882–2895. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, W.; Zhao, C.; Shi, R.; Xue, R.; Li, X. Revegetation by sowing reduces soil bacterial and fungal diversity. Ecol. Evol. 2019, 10, 431–440. [Google Scholar] [CrossRef]
- Njeru, E.M.; Avio, L.; Bocci, G.; Sbrana, C.; Turrini, A.; Bàrberi, P.; Giovannetti, M.; Oehl, F. Contrasting effects of cover crops on ‘hot spot’ arbuscular mycorrhizal fungal communities in organic tomato. Biol. Fertil. Soils 2015, 51, 151–166. [Google Scholar] [CrossRef]
- Johnson, D.; Vandenkoornhuyse, P.J.; Leake, J.R.; Gilbert, L.; Booth, R.E.; Grime, J.P.; Young, J.P.W.; Read, D.J. Plant communities affect arbuscular mycorrhizal fungal diversity and community composition in grassland microcosms. New Phytol. 2003, 161, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Husband, R.; Herre, E.A.; Turner, S.L.; Gallery, R.; Young, J.P.W. Molecular diversity of arbuscular mycorrhizal fungi and patterns of host association over time and space in a tropical forest. Mol. Ecol. 2002, 11, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Helgason, T.; Merryweather, J.W.; Denison, J.; Wilson, P.; Young, J.P.W.; Fitter, A.H. Selectivity and functional diversity in arbuscular mycorrhizas of co-occurring fungi and plants from a temperate deciduous woodland. J. Ecol. 2002, 90, 371–384. [Google Scholar] [CrossRef]
- Badri, A.; Stefani, F.O.P.; Lachance, G.; Roy-Arcand, L.; Beaudet, D.; Vialle, A.; Hijri, M. Molecular diagnostic toolkit for Rhizophagus irregularis isolate DAOM-197198 using quantitative PCR assay targeting the mitochondrial genome. Mycorrhiza 2016, 26, 721–733. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raveau, R.; Lounès-Hadj Sahraoui, A.; Hijri, M.; Fontaine, J. Clary Sage Cultivation and Mycorrhizal Inoculation Influence the Rhizosphere Fungal Community of an Aged Trace-Element Polluted Soil. Microorganisms 2021, 9, 1333. https://doi.org/10.3390/microorganisms9061333
Raveau R, Lounès-Hadj Sahraoui A, Hijri M, Fontaine J. Clary Sage Cultivation and Mycorrhizal Inoculation Influence the Rhizosphere Fungal Community of an Aged Trace-Element Polluted Soil. Microorganisms. 2021; 9(6):1333. https://doi.org/10.3390/microorganisms9061333
Chicago/Turabian StyleRaveau, Robin, Anissa Lounès-Hadj Sahraoui, Mohamed Hijri, and Joël Fontaine. 2021. "Clary Sage Cultivation and Mycorrhizal Inoculation Influence the Rhizosphere Fungal Community of an Aged Trace-Element Polluted Soil" Microorganisms 9, no. 6: 1333. https://doi.org/10.3390/microorganisms9061333
APA StyleRaveau, R., Lounès-Hadj Sahraoui, A., Hijri, M., & Fontaine, J. (2021). Clary Sage Cultivation and Mycorrhizal Inoculation Influence the Rhizosphere Fungal Community of an Aged Trace-Element Polluted Soil. Microorganisms, 9(6), 1333. https://doi.org/10.3390/microorganisms9061333